

Alagille syndrome: pathogenesis, diagnosis and management
- PMID: 21934706
- PMCID: PMC3283172
- DOI: 10.1038/ejhg.2011.181
Alagille syndrome: pathogenesis, diagnosis and management
Abstract
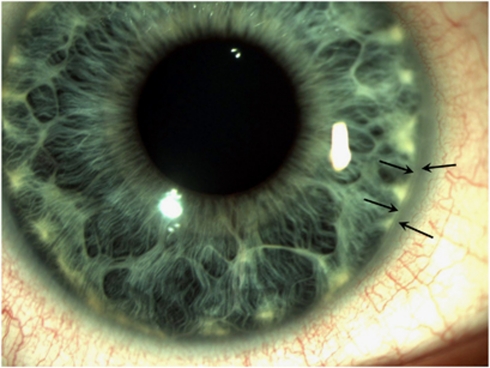
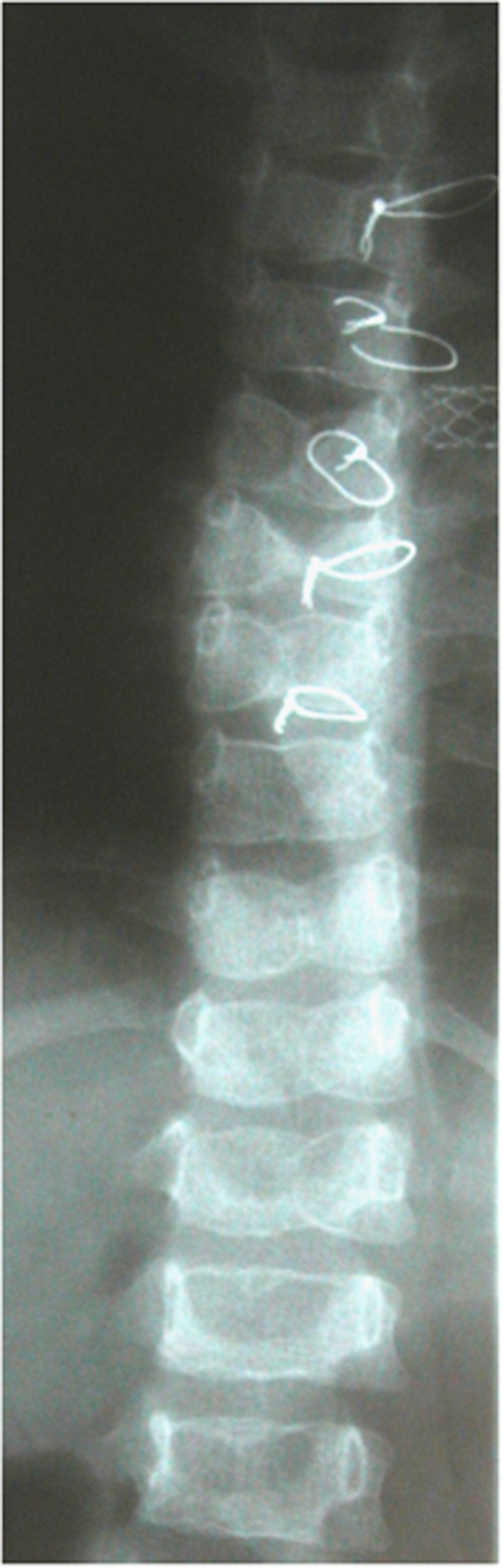
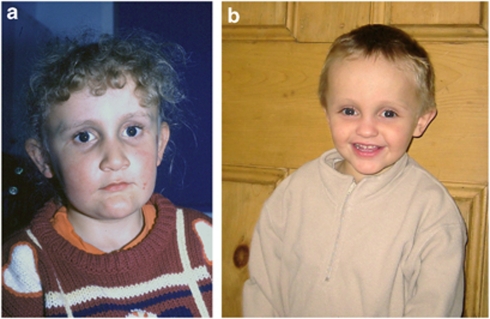
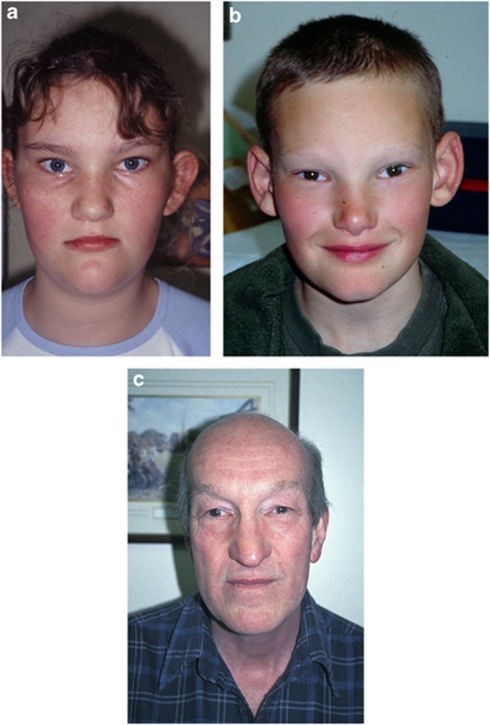
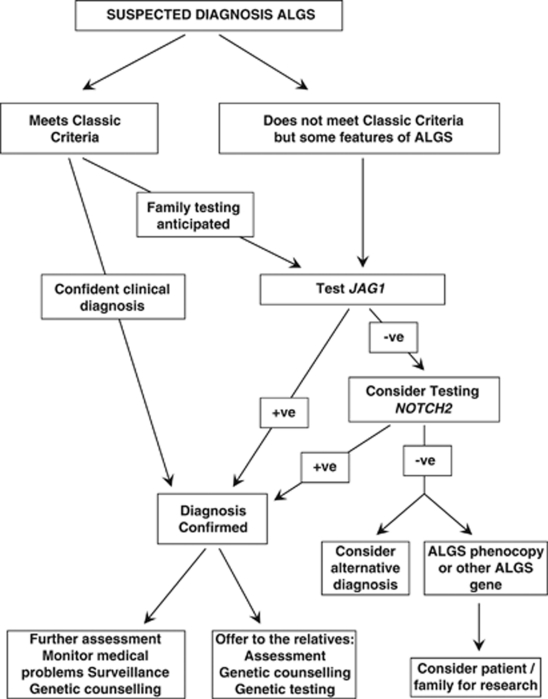
Alagille syndrome (ALGS), also known as arteriohepatic dysplasia, is a multisystem disorder due to defects in components of the Notch signalling pathway, most commonly due to mutation in JAG1 (ALGS type 1), but in a small proportion of cases mutation in NOTCH2 (ALGS type 2). The main clinical and pathological features are chronic cholestasis due to paucity of intrahepatic bile ducts, peripheral pulmonary artery stenosis, minor vertebral segmentation anomalies, characteristic facies, posterior embryotoxon/anterior segment abnormalities, pigmentary retinopathy, and dysplastic kidneys. It follows autosomal dominant inheritance, but reduced penetrance and variable expression are common in this disorder, and somatic/germline mosaicism may also be relatively frequent. This review discusses the clinical features of ALGS, including long-term complications, the clinical and molecular diagnosis, and management.
Figures

References
-
- Alagille D, Habib EC, Thomassin N. Editions Medicales Flammarion, Paris; 1969. L'atresie des voies biliaires intrahepatiques avec voies biliaires extrahepatiques permeables chez l'enfant; pp. 301–318.
-
- Alagille D, Odièvre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. J Pediatr. 1975;86:63–71. - PubMed
-
- Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
