

Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells
- PMID: 21940781
- PMCID: PMC3198104
- DOI: 10.2337/db11-0341
Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells
Abstract
Objective: Insulin receptor substrate-2 (IRS-2) plays an essential role in pancreatic islet β-cells by promoting growth and survival. IRS-2 turnover is rapid in primary β-cells, but its expression is highly regulated at the transcriptional level, especially by glucose. The aim was to investigate the molecular mechanism on how glucose regulates IRS-2 gene expression in β-cells.
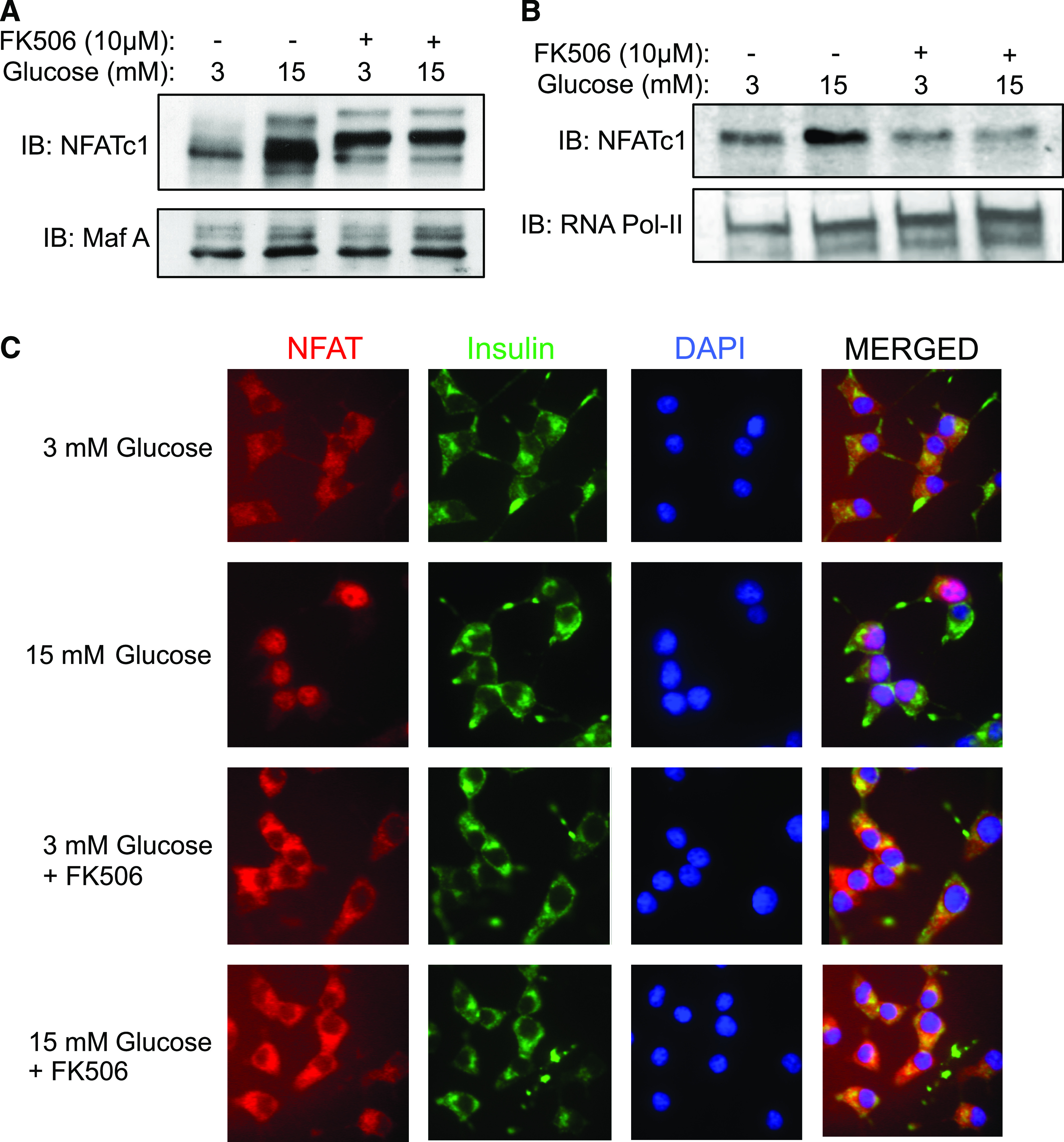
Research design and methods: Rat islets were exposed to inhibitors or subjected to adenoviral vector-mediated gene manipulations and then to glucose-induced IRS-2 expression analyzed by real-time PCR and immunoblotting. Transcription factor nuclear factor of activated T cells (NFAT) interaction with IRS-2 promoter was analyzed by chromatin immunoprecipitation assay and glucose-induced NFAT translocation by immunohistochemistry.
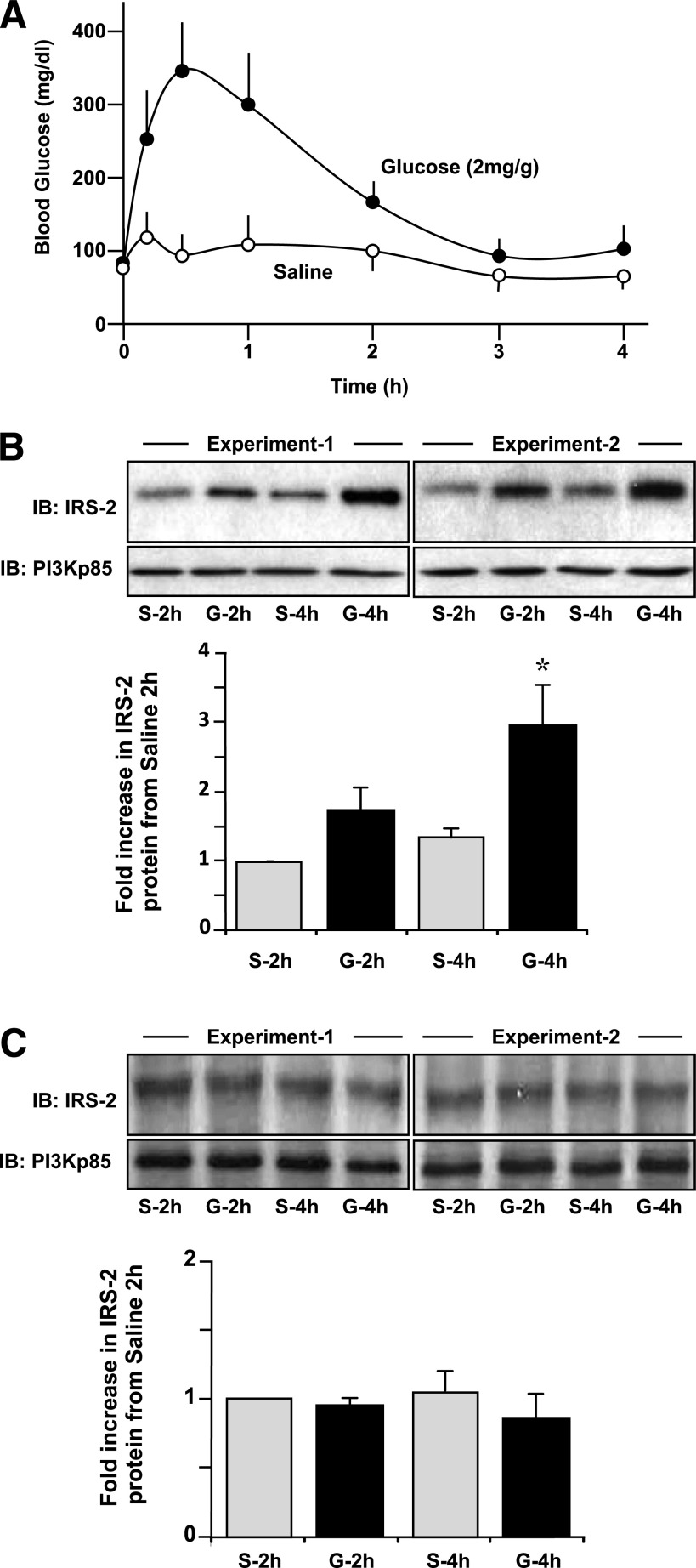
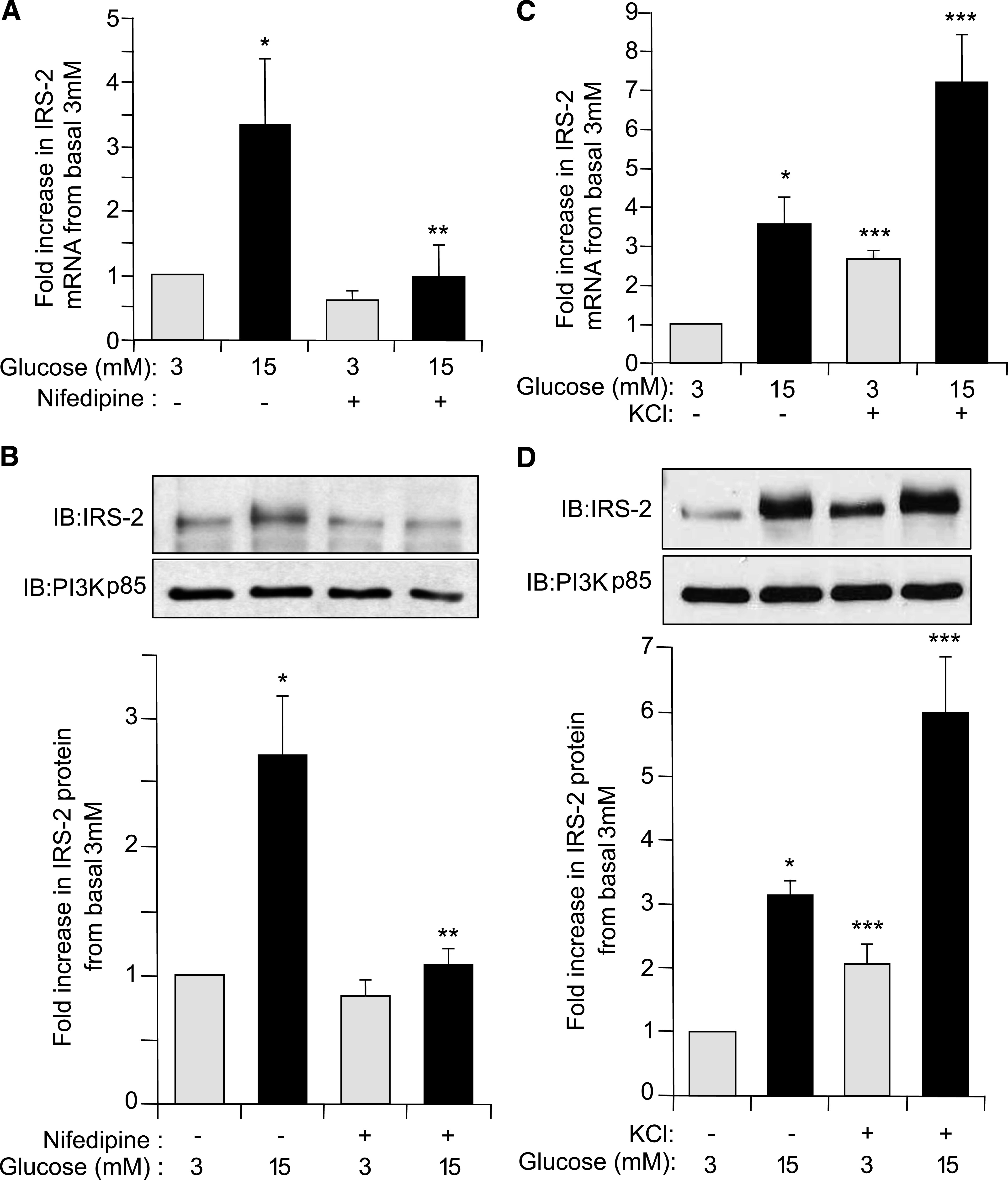
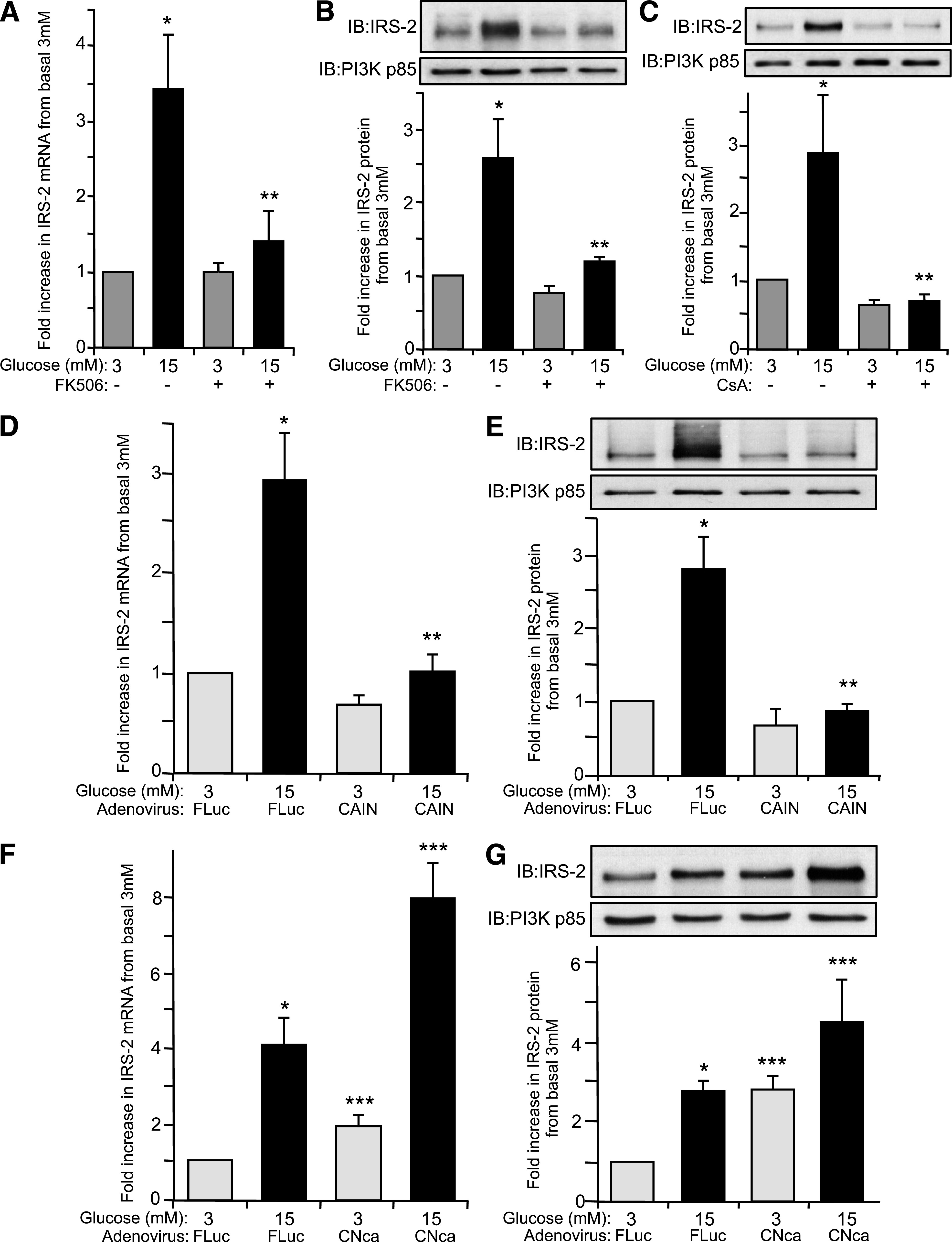
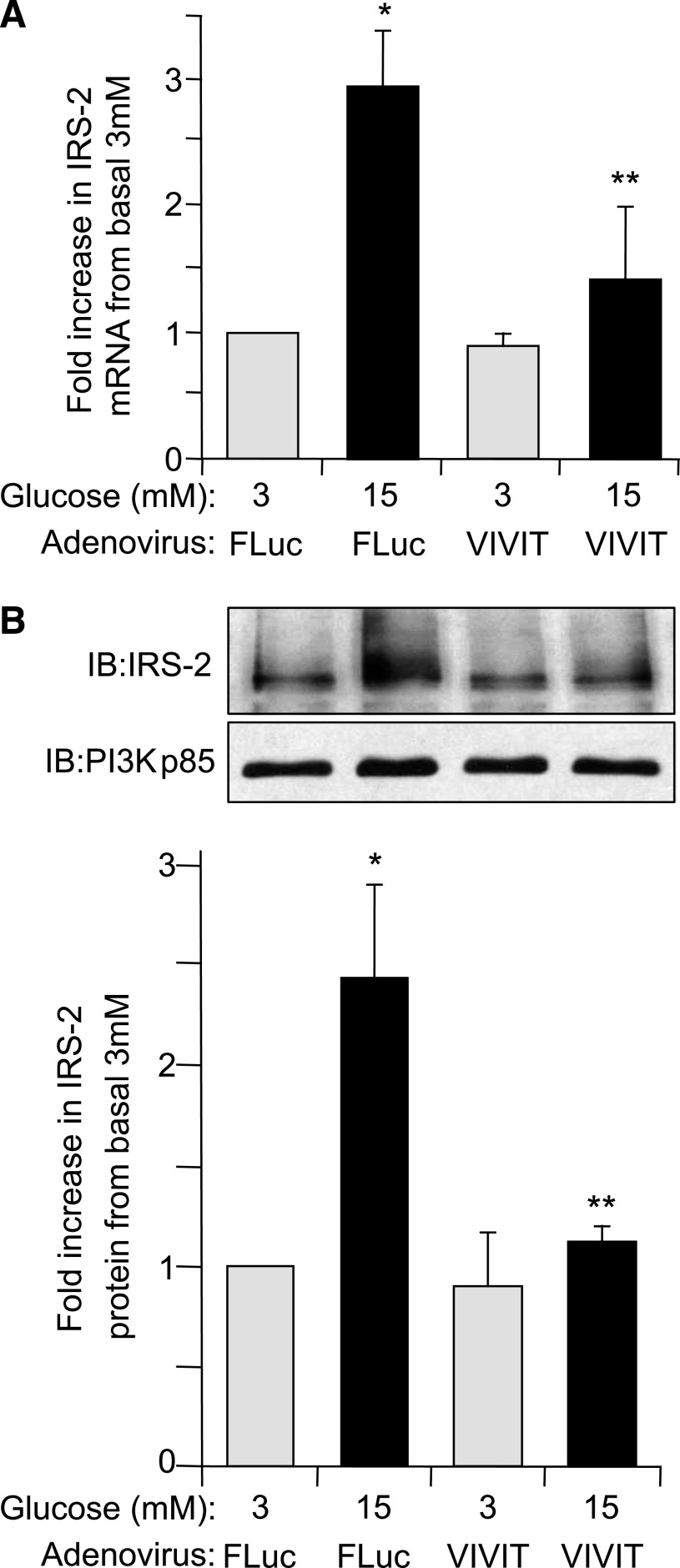
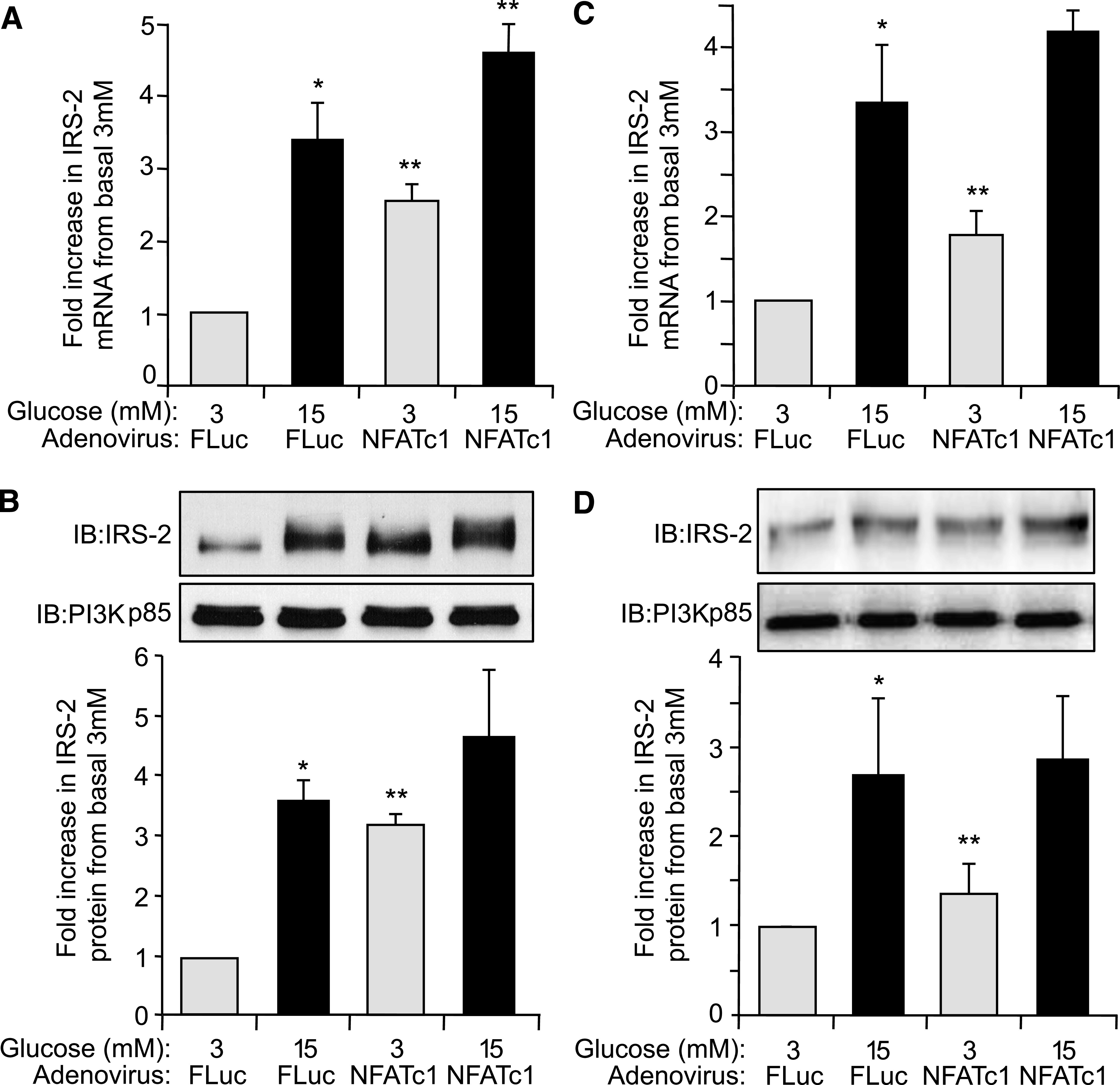
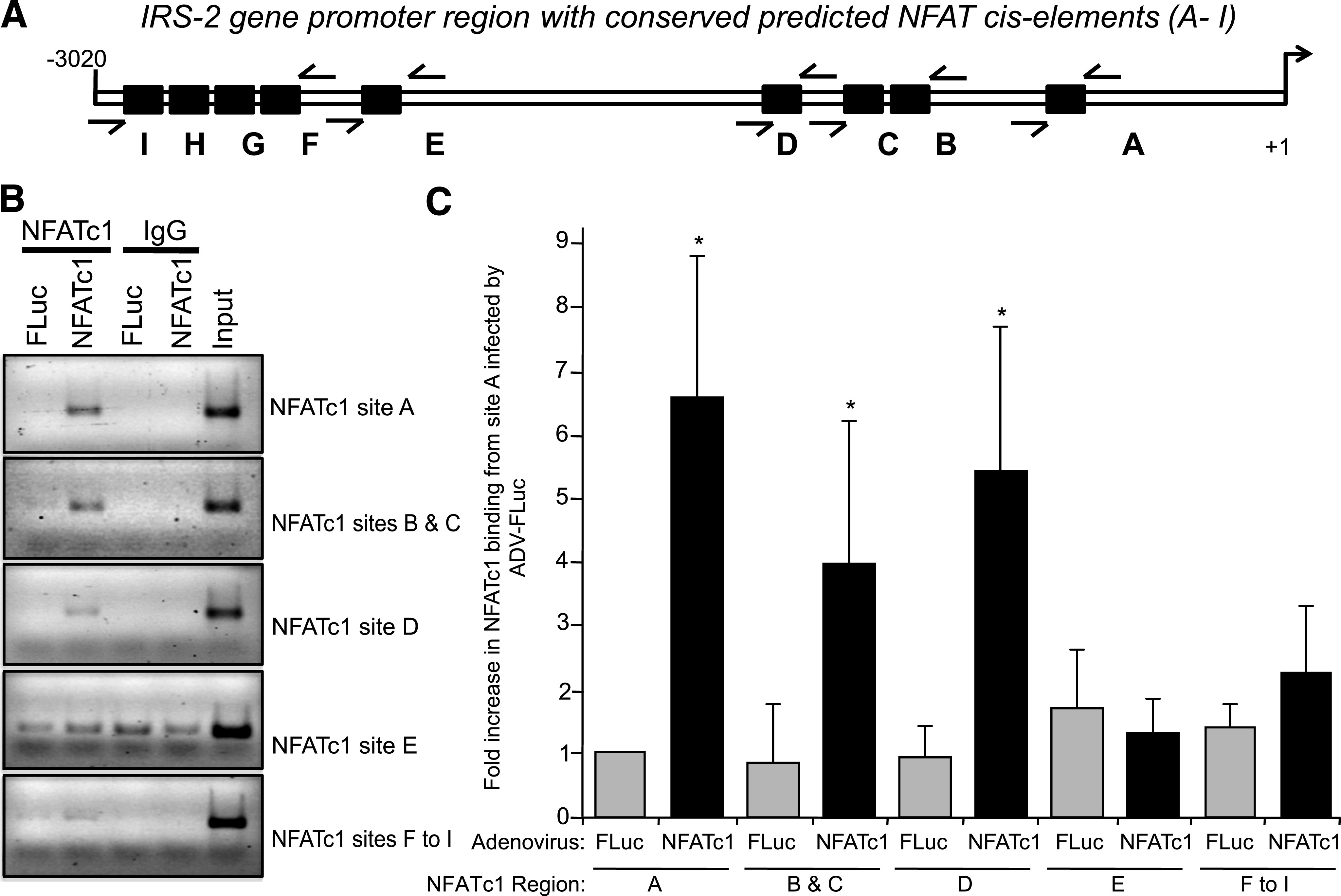
Results: Glucose-induced IRS-2 expression occurred in pancreatic islet β-cells in vivo but not in liver. Modulating rat islet β-cell Ca(2+) influx with nifedipine or depolarization demonstrated that glucose-induced IRS-2 gene expression was dependent on a rise in intracellular calcium concentration derived from extracellular sources. Calcineurin inhibitors (FK506, cyclosporin A, and a peptide calcineurin inhibitor [CAIN]) abolished glucose-induced IRS-2 mRNA and protein levels, whereas expression of a constitutively active calcineurin increased them. Specific inhibition of NFAT with the peptide inhibitor VIVIT prevented a glucose-induced IRS-2 transcription. NFATc1 translocation to the nucleus in response to glucose and association of NFATc1 to conserved NFAT binding sites in the IRS-2 promoter were demonstrated.
Conclusions: The mechanism behind glucose-induced transcriptional control of IRS-2 gene expression specific to the islet β-cell is mediated by the Ca(2+)/calcineurin/NFAT pathway. This insight into the IRS-2 regulation could provide novel therapeutic means in type 2 diabetes to maintain an adequate functional mass.
Figures
References
-
- Rhodes CJ. Type 2 diabetes-a matter of β-cell life and death? Science 2005;307:380–384 - PubMed
-
- Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab 2007;3:758–768 - PubMed
-
- Withers DJ, Gutierrez JS, Towery H, et al. . Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–904 - PubMed
-
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 1999;23:32–40 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
