

New molecular targets in mantle cell lymphoma
- PMID: 21945517
- PMCID: PMC3217176
- DOI: 10.1016/j.semcancer.2011.09.008
New molecular targets in mantle cell lymphoma
Abstract
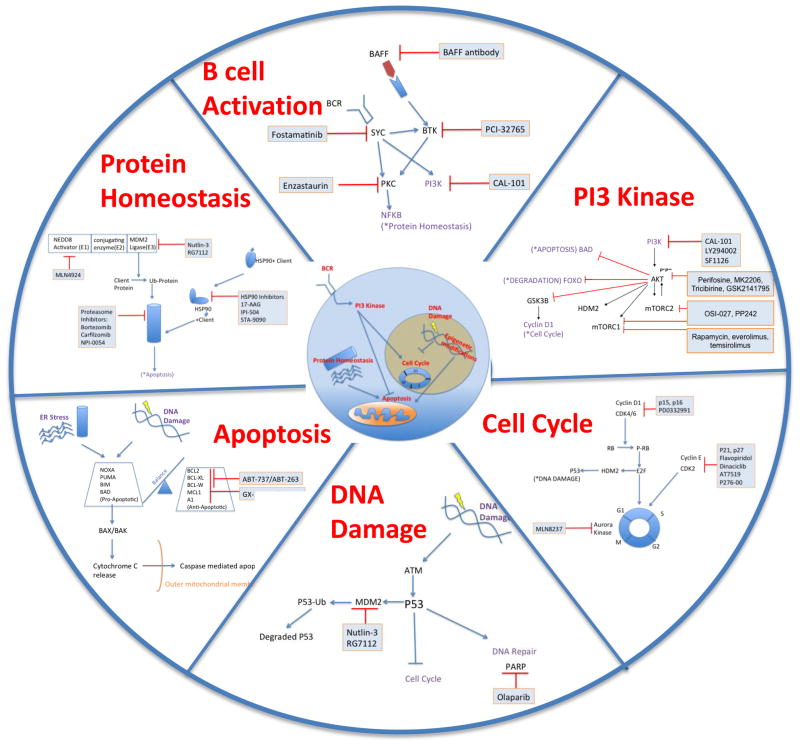
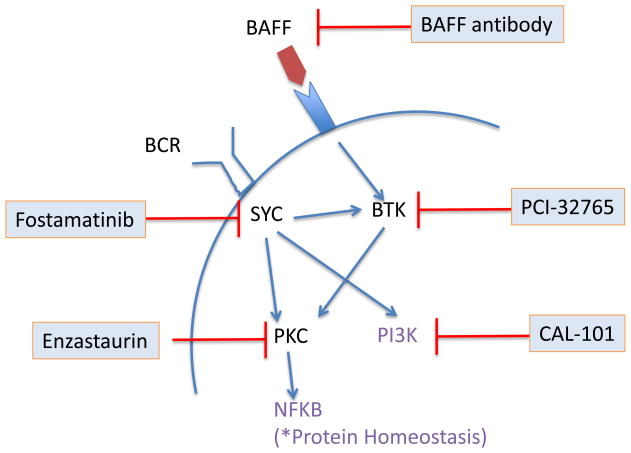
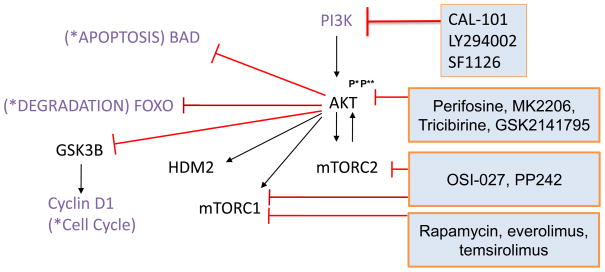
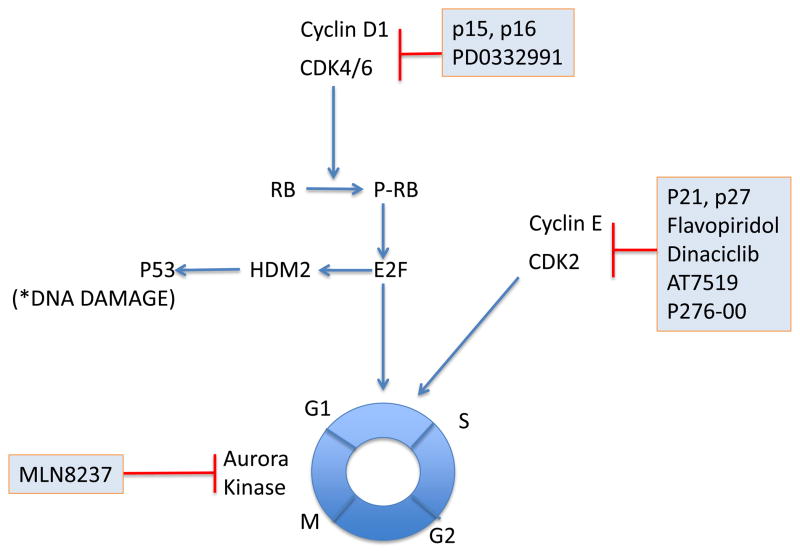
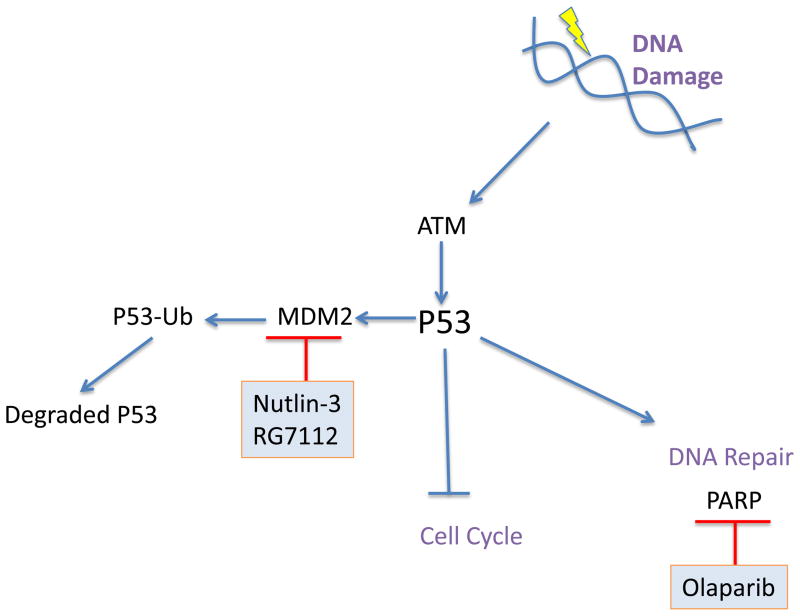
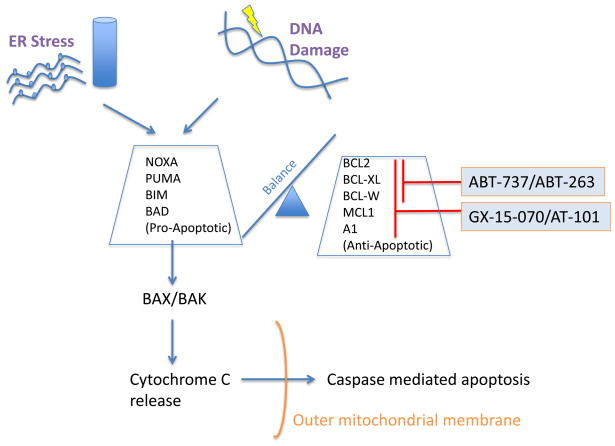
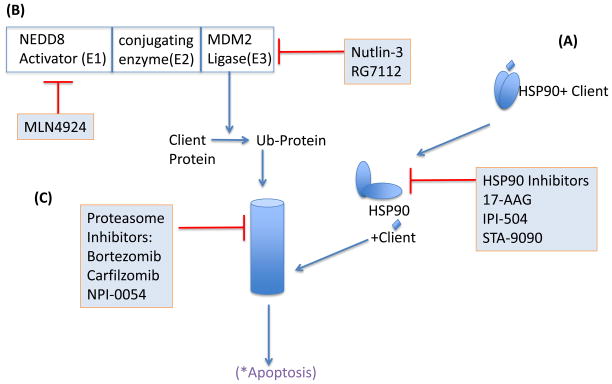
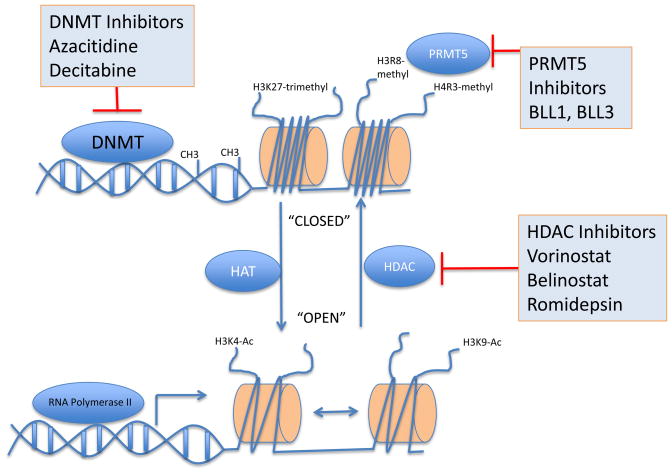
Mantle cell lymphoma (MCL) is a malignancy of mature B cells characterized by aberrant expression of cyclin D1 due to the translocation t(11;14). Epigenomic and genomic lesions in pathways regulating B-cell activation, cell cycle progression, protein homeostasis, DNA damage response, cell proliferation and apoptosis contribute to its pathogenesis. While patients typically respond to first-line chemotherapy, relapse is the rule resulting in a median survival of 5-7 years. The PI3K/AKT/mTOR appears as a key pathway in the pathogenesis and can be targeted with small molecules. Most experience is with mTOR inhibitors of the rapamycin class. Second-generation mTOR inhibitors and the PI3K inhibitor CAL-101 are novel options to more effectively target this pathway. Bruton's tyrosine kinase inhibition by PCI-32765 has promising activity and indicates immunoreceptor signaling as a novel therapeutic target. Up to 50% of relapsed patients respond to the proteasome inhibitor bortezomib suggesting that MCL may be particularly sensitive to disruption of protein homeostasis and/or induction of oxidative stress. Recent work has focused on elucidating the mechanism of bortezomib-induced cytotoxicity and the development of second-generation proteasome inhibitors. DNA hypomethylating agents and histone deacetylase inhibitors effect epigenetic de-repression of aberrantly silenced genes. These epigenetic pharmaceuticals and HSP90 inhibitors can synergize with proteasome inhibitors. Finally, BH3 mimetics are emerging as tools to sensitize tumor cells to chemotherapy. Participation in clinical trials offers patients a chance to benefit from these advances and is essential to maintain the momentum of progress. Innovative trial designs may be needed to expedite the clinical development of these targeted agents.
Published by Elsevier Ltd.
Figures
References
-
- Rinaldi A, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. British journal of haematology. 2006;132:303–316. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
