

Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155
- PMID: 21946536
- PMCID: PMC3501198
- DOI: 10.1038/nm.2459
Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155
Erratum in
- Nat Med. 2011 Nov;17(11):1521
- Nat Med. 2011 Oct;17(10):2 p following 1282
Abstract
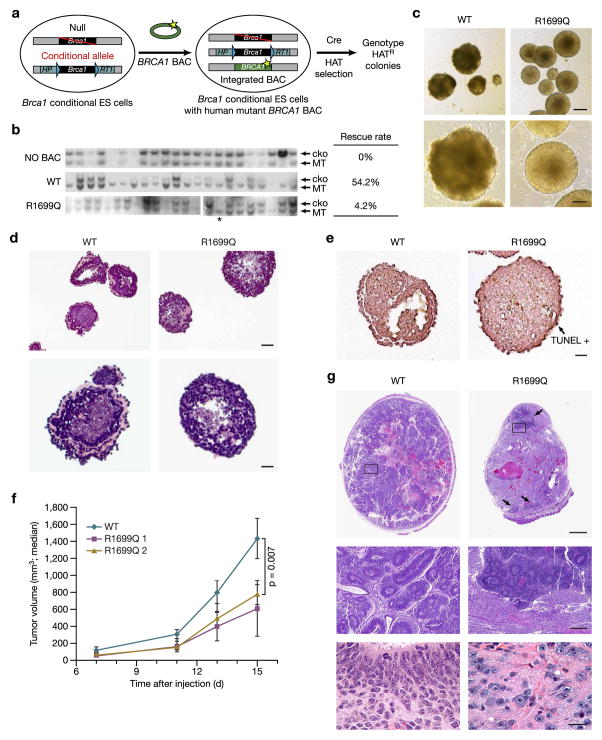
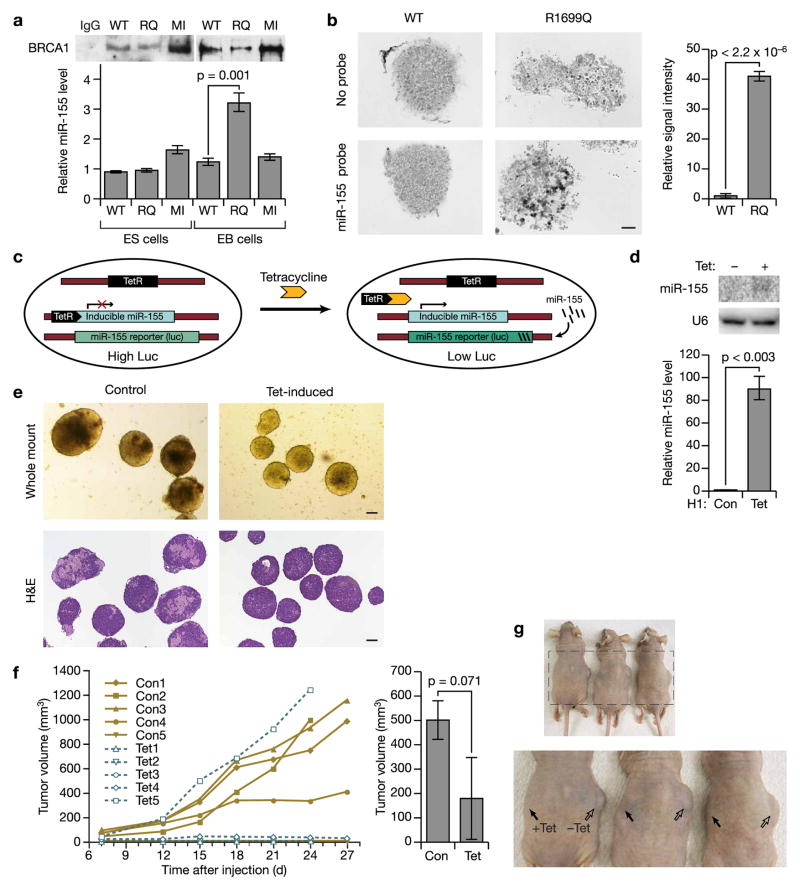
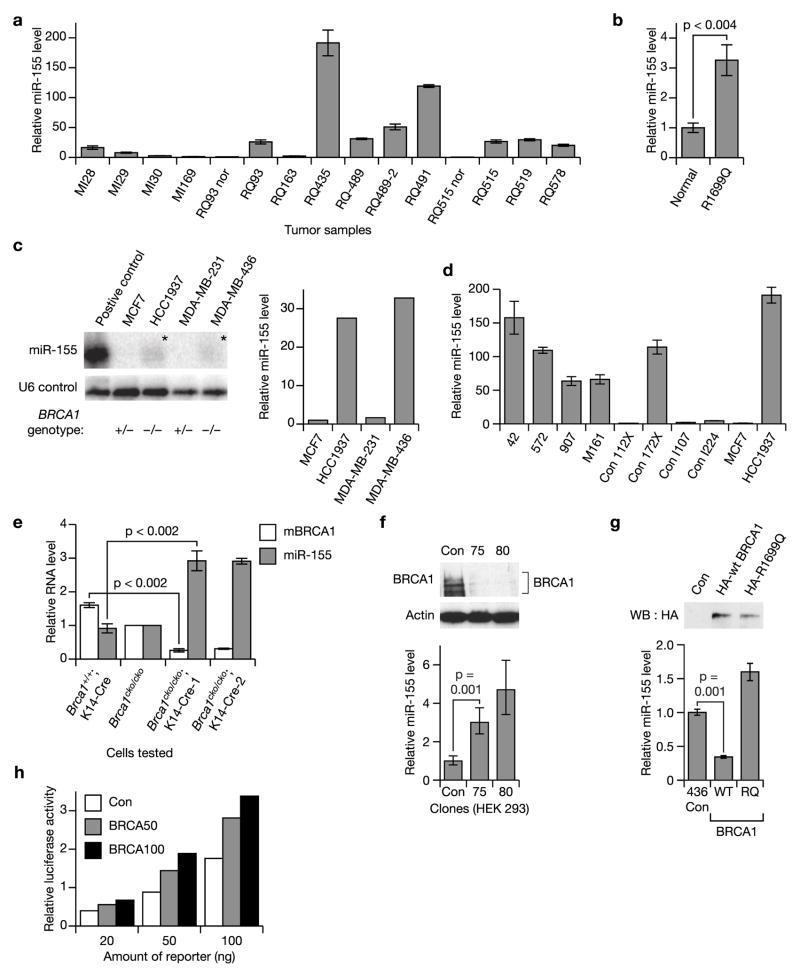
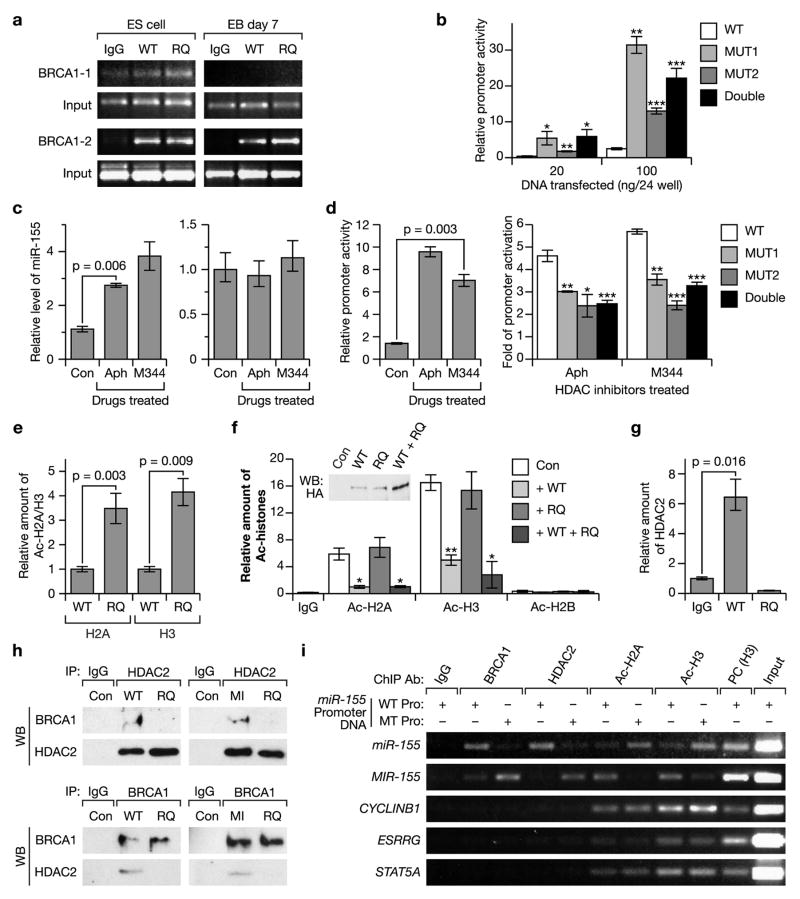
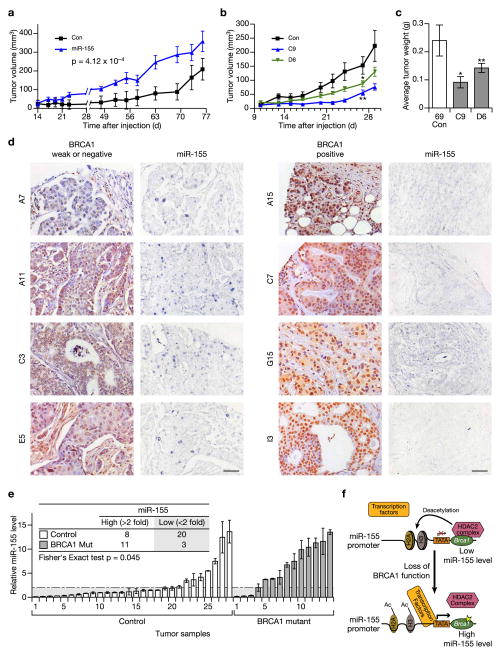
BRCA1, a well-known tumor suppressor with multiple interacting partners, is predicted to have diverse biological functions. However, so far its only well-established role is in the repair of damaged DNA and cell cycle regulation. In this regard, the etiopathological study of low-penetrant variants of BRCA1 provides an opportunity to uncover its other physiologically important functions. Using this rationale, we studied the R1699Q variant of BRCA1, a potentially moderate-risk variant, and found that it does not impair DNA damage repair but abrogates the repression of microRNA-155 (miR-155), a bona fide oncomir. Mechanistically, we found that BRCA1 epigenetically represses miR-155 expression via its association with HDAC2, which deacetylates histones H2A and H3 on the miR-155 promoter. We show that overexpression of miR-155 accelerates but the knockdown of miR-155 attenuates the growth of tumor cell lines in vivo. Our findings demonstrate a new mode of tumor suppression by BRCA1 and suggest that miR-155 is a potential therapeutic target for BRCA1-deficient tumors.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Comment in
-
Epigenetic tumor suppression by BRCA1.Nat Med. 2011 Oct 11;17(10):1183-5. doi: 10.1038/nm.2493. Nat Med. 2011. PMID: 21988990 No abstract available.
References
-
- O’Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31:961–967. - PubMed
-
- Venkitaraman AR. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu Rev Pathol. 2009;4:461–487. - PubMed
-
- Xia Y, Pao GM, Chen HW, Verma IM, Hunter T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem. 2003;278:5255–5263. - PubMed
-
- Baer R, Ludwig T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev. 2002;12:86–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
