

Review
doi: 10.1007/s00467-011-1992-9.
Epub 2011 Sep 27.
Tubulointerstitial injury and the progression of chronic kidney disease
Affiliations
- PMID: 21947270
- PMCID: PMC3337413
- DOI: 10.1007/s00467-011-1992-9
Item in Clipboard
Review
Tubulointerstitial injury and the progression of chronic kidney disease
Pediatr Nephrol.
2012 Jun.
Abstract
In chronic kidney disease (CKD), once injury from any number of disease processes reaches a threshold, there follows an apparently irreversible course toward decline in kidney function. The tubulointerstitium may play a key role in this common progression pathway. Direct injury, high metabolic demands, or stimuli from various other forms of renal dysfunction activate tubular cells. These, in turn, interact with interstitial tissue elements and inflammatory cells, causing further pathologic changes in the renal parenchyma. The tissue response to these changes thus generates a feed-forward loop of kidney injury and progressive loss of function. This article reviews the mechanisms of this negative cycle mediating CKD.
Figures

Glomerular or tubular injury initiates chronic kidney disease (CKD), but the tubulointerstitial response is primarily responsible for progression. (1) Podocyte dysfunction or depletion leads to proteinuria, causing reactive changes in the tubular cells. (2) Podocyte depletion also permits the transudation of glomerular filtrate directly into the periglomerular tubulointerstitium, depositing a number of biologically active molecules in that compartment. (3) As the number of effective nephrons decreases, the remaining nephrons hypertrophy. An increased amount of filtrate per nephron increases metabolic demand on the tubules, which under even normal circumstances have high energy requirements. Thus, further stress on tubule cell homeostasis results in activation of the cells, away from a state of relatively orderly transporter function. ROS Reactive oxygen species
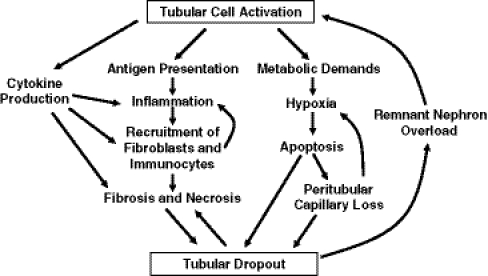
Activated tubular cells participate in a number of processes that lead to progressive glomerular loss. While it is not clear that the cells directly produce extracellular matrix to yield a scar, they do produce cytokines, support inflammatory responses, and dedifferentiate or die. The result is nephron loss
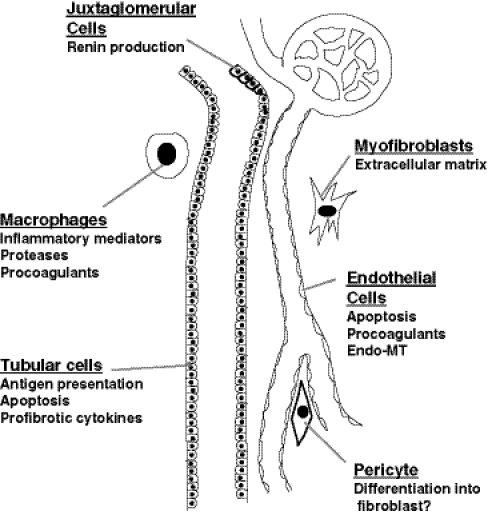
Different cell types contribute to progressive nephron loss. Juxtaglomerular cells produce renin, affecting tubuloglomerular feedback to alter glomerular filtration and activating extracellular matrix (ECM) production by multiple cell types. Tubular cells dedifferentiate to recruit inflammatory cells and activated fibroblasts. These myofibroblasts may be derived from vascular pericytes, circulating or resident fibroblasts, or endothelial cells. Macrophages also promote inflammation and tissue damage through protease production. Endothelial cell apoptosis accelerates hypoxic and ischemic changes of the tubular and interstitial cells. The production of soluble mediators by these cells permits them to interact in a way that under ideal circumstances orchestrates a healing response, but in CKD promotes a vicious cycle of misdirected repair

The cycle of dysfunction in progressive CKD. Decreased glomerular filtration rate (GFR) or primary tubulointerstitial damage leads to poorly functioning glomeruli and the deleterious activation of tubular cells. These cells communicate with other cells to orchestrate a series of events that trigger mechanisms of interstitial inflammation, fibrosis, and cell death. The result is loss of peritubular capillaries, tubular cell apoptosis and necrosis, and nephron loss. Hypertrophic responses to these events further damage the remnant glomeruli and tubules, propagating the cycle of injury and misdirected repair. RAS Renin–angiotensin–aldosterone system, EMT epithelial-to-mesenchymal transition
References
-
- Bohle A, Mackensen-Haen S, VonGise, Grund KE, Wehrmann M, Batz C, Bogen Schutz O, Schmitt H, Nagy J, Muller C. The consequences of tubule-interstitial changes for renal function in glomerulopathies. A morphometric and cytological analysis. Pathol Res Pract. 1990;186:135–144. doi: 10.1016/S0344-0338(11)81021-6. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
