

HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response
- PMID: 21949742
- PMCID: PMC3176279
- DOI: 10.1371/journal.pone.0024660
HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response
Abstract
Background: The endoplasmic reticulum (ER) is the cellular site for protein folding. ER stress occurs when protein folding capacity is exceeded. This stress induces a cyto-protective signaling cascades termed the unfolded protein response (UPR) aimed at restoring homeostasis. While acute ER stress is lethal, chronic sub-lethal ER stress causes cells to adapt by attenuation of UPR activation. Hepatitis C virus (HCV), a major human pathogen, was shown to cause ER stress, however it is unclear whether HCV induces chronic ER stress, and if so whether adaptation mechanisms are initiated. We wanted to characterize the kinetics of HCV-induced ER stress during infection and assess adaptation mechanisms and their significance.
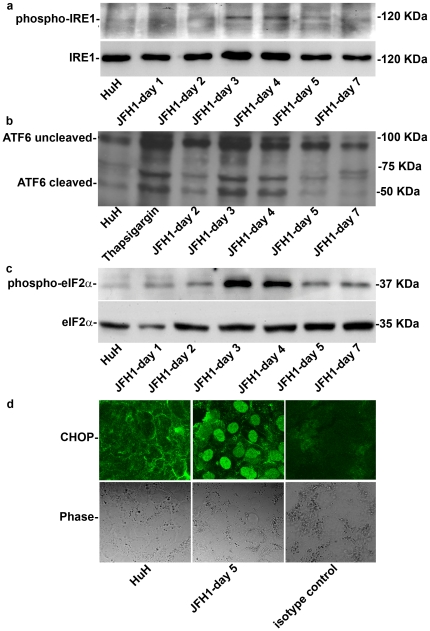
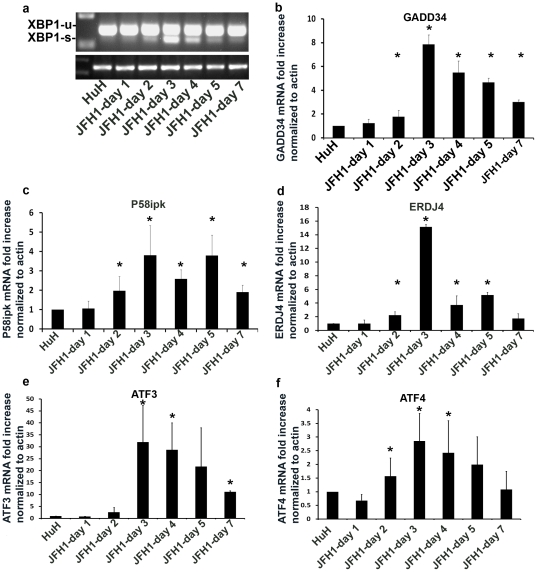
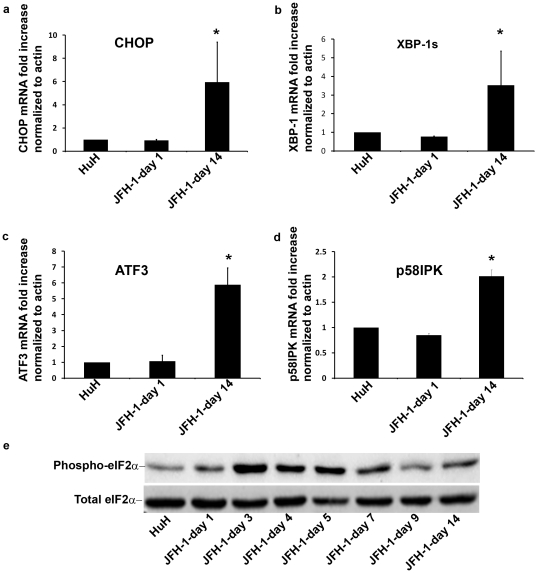
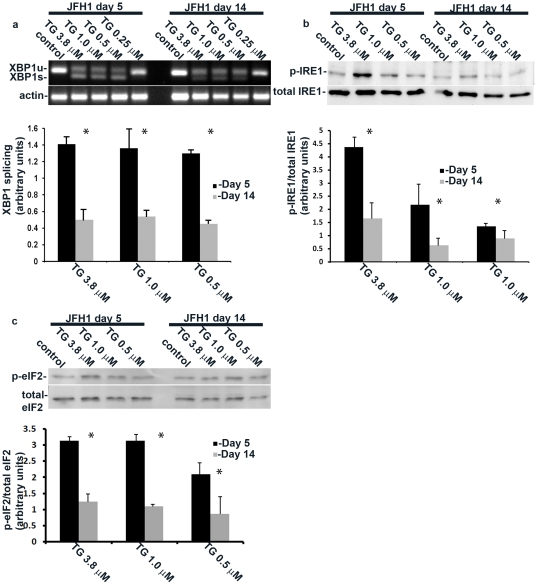
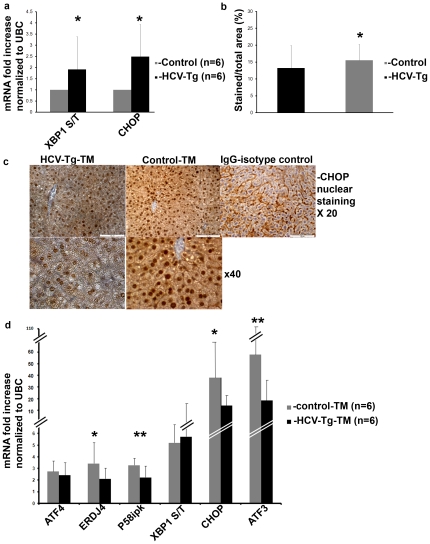
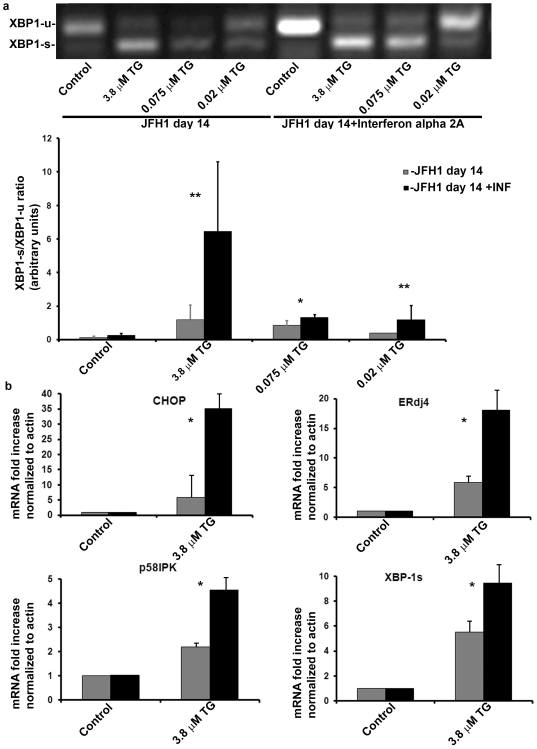
Methods and findings: The HuH7.5.1 cellular system and HCV-transgenic (HCV-Tg) mice were used to characterize HCV-induced ER stress/UPR pathway activation and adaptation. HCV induced a wave of acute ER stress peaking 2-5 days post-infection, which rapidly subsided thereafter. UPR pathways were activated including IRE1 and EIF2α phosphorylation, ATF6 cleavage and XBP-1 splicing. Downstream target genes including GADD34, ERdj4, p58ipk, ATF3 and ATF4 were upregulated. CHOP, a UPR regulated protein was activated and translocated to the nucleus. Remarkably, UPR activity did not return to baseline but remained elevated for up to 14 days post infection suggesting that chronic ER stress is induced. At this time, cells adapted to ER stress and were less responsive to further drug-induced ER stress. Similar results were obtained in HCV-Tg mice. Suppression of HCV by Interferon-α 2a treatment, restored UPR responsiveness to ER stress tolerant cells.
Conclusions: Our study shows, for the first time, that HCV induces adaptation to chronic ER stress which was reversed upon viral suppression. These finding represent a novel viral mechanism to manipulate cellular response pathways.
Conflict of interest statement
Figures
References
-
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558–67. - PubMed
-
- Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436(7053):939–45. - PubMed
-
- Pereira AA, Jacobson IM. New and experimental therapies for HCV. Nat Rev Gastroenterol Hepatol. 2009;6(7):403–11. - PubMed
-
- Dubuisson J, Penin F, Moradpour D. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol. 2002;12(11):517–23. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
