

Rapid genotyping of soybean cultivars using high throughput sequencing
- PMID: 21949759
- PMCID: PMC3176760
- DOI: 10.1371/journal.pone.0024811
Rapid genotyping of soybean cultivars using high throughput sequencing
Abstract
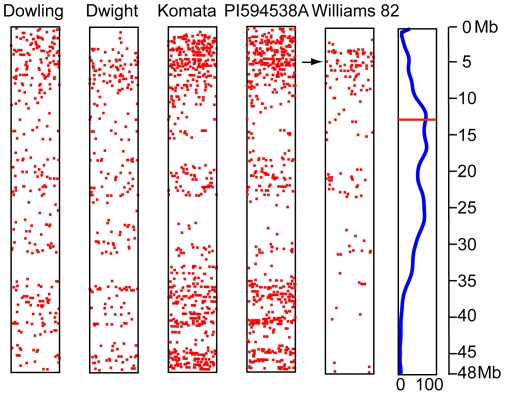
Soybean (Glycine max) breeding involves improving commercially grown varieties by introgressing important agronomic traits from poor yielding accessions and/or wild relatives of soybean while minimizing the associated yield drag. Molecular markers associated with these traits are instrumental in increasing the efficiency of producing such crosses and Single Nucleotide Polymorphisms (SNPs) are particularly well suited for this task, owing to high density in the non-genic regions and thus increased likelihood of finding a tightly linked marker to a given trait. A rapid method to develop SNP markers that can differentiate specific loci between any two parents in soybean is thus highly desirable. In this study we investigate such a protocol for developing SNP markers between multiple soybean accessions and the reference Williams 82 genome. To restrict sampling frequency reduced representation libraries (RRLs) of genomic DNA were generated by restriction digestion followed by library construction. We chose to sequence four accessions Dowling (PI 548663), Dwight (PI 597386), Komata (PI200492) and PI 594538A for their agronomic importance as well as Williams 82 as a control.MseI was chosen to digest genomic DNA based on predictions that it will cut sparingly in the mathematically defined high-copy-number regions of the genome. All RRLs were sequenced on the Illumina genome analyzer. Reads were aligned to the Glyma1 reference assembly and SNP calls made from the alignments. We identified from 4294 to 14550 SNPs between the four accessions and the Williams 82 reference. In addition a small number of SNPs (1142) were found by aligning Williams 82 reads to the reference assembly (Glyma1) suggesting limited genetic variation within the Williams 82 line. The SNP data allowed us to estimate genetic diversity between the four lines and Williams 82. Restriction digestion of soybean genomic DNA with MseI followed by high throughput sequencing provides a rapid and reproducible method for generating SNP markers.
Conflict of interest statement
Figures



 = 3 reads in 1–5 lines are shown.
= 3 reads in 1–5 lines are shown.


References
-
- Bernard R, Cremeens C. Registration of ‘Williams 82’ soybean. Crop Science. 1988;28:1027–1028.
-
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. - PubMed
-
- Keim P, Shoemaker RC, Palmer RG. Restriction fragment length polymor- phism diversity in soybean. TAG Theoretical and Applied Genetics. 1989;77:786–792. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
