

Evaluation of intra-host variants of the entire hepatitis B virus genome
- PMID: 21949887
- PMCID: PMC3176825
- DOI: 10.1371/journal.pone.0025232
Evaluation of intra-host variants of the entire hepatitis B virus genome
Abstract
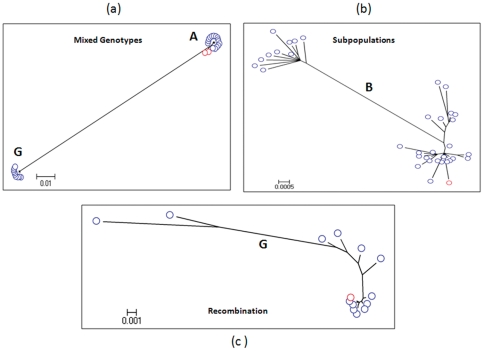
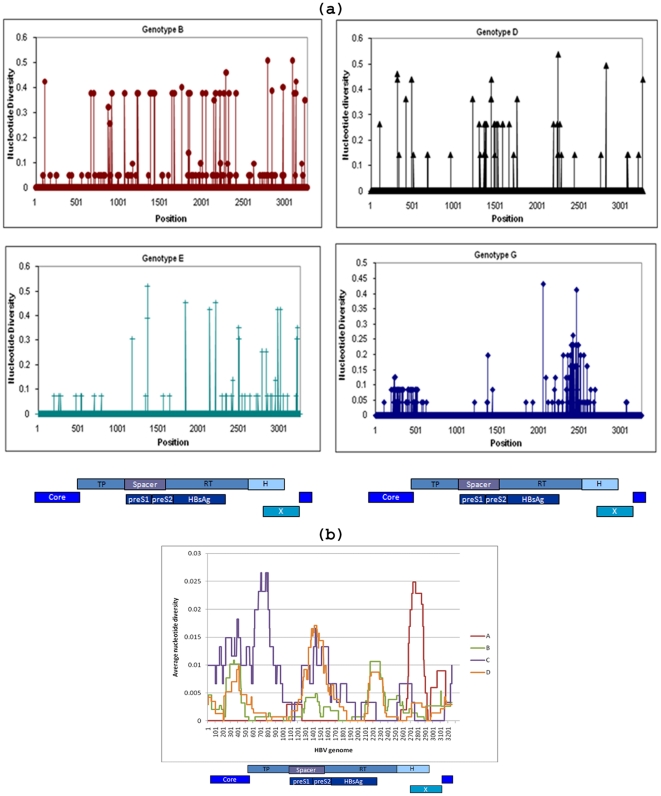
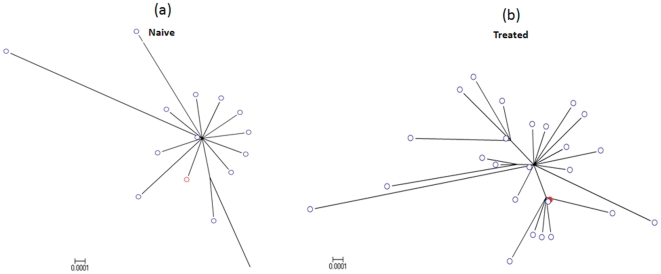
Genetic analysis of hepatitis B virus (HBV) frequently involves study of intra-host variants, identification of which is commonly achieved using short regions of the HBV genome. However, the use of short sequences significantly limits evaluation of genetic relatedness among HBV strains. Although analysis of HBV complete genomes using genetic cloning has been developed, its application is highly labor intensive and practiced only infrequently. We describe here a novel approach to whole genome (WG) HBV quasispecies analysis based on end-point, limiting-dilution real-time PCR (EPLD-PCR) for amplification of single HBV genome variants, and their subsequent sequencing. EPLD-PCR was used to analyze WG quasispecies from serum samples of patients (n = 38) infected with HBV genotypes A, B, C, D, E and G. Phylogenetic analysis of the EPLD-isolated HBV-WG quasispecies showed the presence of mixed genotypes, recombinant variants and sub-populations of the virus. A critical observation was that HBV-WG consensus sequences obtained by direct sequencing of PCR fragments without EPLD are genetically close, but not always identical to the major HBV variants in the intra-host population, thus indicating that consensus sequences should be judiciously used in genetic analysis. Sequence-based studies of HBV WG quasispecies should afford a more accurate assessment of HBV evolution in various clinical and epidemiological settings.
Conflict of interest statement
Figures

Similar articles
-
Hepatitis B virus genomic heterogeneity: variation between quasispecies may confound molecular epidemiological analyses of transmission incidents.J Viral Hepat. 1997 Sep;4(5):309-15. doi: 10.1046/j.1365-2893.1997.00066.x. J Viral Hepat. 1997. PMID: 9310929
-
Epidemic history and evolutionary dynamics of hepatitis B virus infection in two remote communities in rural Nigeria.PLoS One. 2010 Jul 19;5(7):e11615. doi: 10.1371/journal.pone.0011615. PLoS One. 2010. PMID: 20657838 Free PMC article.
-
Phylogenetic analysis of complete genome sequences of hepatitis B virus from an Afro-Colombian community: presence of HBV F3/A1 recombinant strain.Virol J. 2012 Oct 24;9:244. doi: 10.1186/1743-422X-9-244. Virol J. 2012. PMID: 23092209 Free PMC article.
-
Hepatitis B virus intergenotypic recombinants worldwide: An overview.Infect Genet Evol. 2015 Dec;36:500-510. doi: 10.1016/j.meegid.2015.08.024. Epub 2015 Aug 20. Infect Genet Evol. 2015. PMID: 26299884 Review.
-
Hepatitis B virus genotypes: comparison of genotyping methods.Rev Med Virol. 2004 Jan-Feb;14(1):3-16. doi: 10.1002/rmv.400. Rev Med Virol. 2004. PMID: 14716688 Review.
Cited by
-
Convergence and coevolution of hepatitis B virus drug resistance.Nat Commun. 2012 Apr 17;3:789. doi: 10.1038/ncomms1794. Nat Commun. 2012. PMID: 22510694 Free PMC article.
-
Integrated HIV surveillance finds recent adult hepatitis B virus (HBV) transmission and intermediate HBV prevalence among military in uncharacterized Caribbean country.PLoS One. 2019 Oct 1;14(10):e0222835. doi: 10.1371/journal.pone.0222835. eCollection 2019. PLoS One. 2019. PMID: 31574098 Free PMC article.
-
Complex genetic encoding of the hepatitis B virus on-drug persistence.Sci Rep. 2020 Sep 23;10(1):15574. doi: 10.1038/s41598-020-72467-9. Sci Rep. 2020. PMID: 32968103 Free PMC article.
-
Viral quasispecies evolution.Microbiol Mol Biol Rev. 2012 Jun;76(2):159-216. doi: 10.1128/MMBR.05023-11. Microbiol Mol Biol Rev. 2012. PMID: 22688811 Free PMC article. Review.
-
Recent population expansions of hepatitis B virus in the United States.J Virol. 2014 Dec;88(24):13971-80. doi: 10.1128/JVI.01594-14. Epub 2014 Sep 3. J Virol. 2014. PMID: 25187549 Free PMC article.
References
-
- Mizokami M, Orito E, Ohba K, Ikeo K, Lau JY, et al. Constrained evolution with respect to gene overlap of hepatitis B virus. J Mol Evol. 1997;44(Suppl 1):S83–90. - PubMed
-
- Zhou Y, Holmes EC. Bayesian estimates of the evolutionary rate and age of hepatitis B virus. J Mol Evol. 2007;65:197–205. - PubMed
-
- Cassino L, Laufer N, Salomon H, Campos R, Quarleri J. Hepatitis B precore/core promoter mutations in isolates from HBV-monoinfected and HBV-HIV coinfected patients: a 3-yr prospective study. J Clin Virol. 2009;46:354–359. - PubMed
-
- Wang F, Wang H, Shen H, Meng C, Weng X, et al. Evolution of hepatitis B virus polymerase mutations in a patient with HBeAg-positive chronic hepatitis B virus treated with sequential monotherapy and add-on nucleoside/nucleotide analogues. Clin Ther. 2009;31:360–366. - PubMed
-
- Khudyakov Y. Coevolution and HBV drug resistance. Antivir Ther. 2010;15:505–515. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
