

Role of endothelin-1 in the hyper-responsiveness to nitrovasodilators following acute NOS inhibition
- PMID: 21951225
- PMCID: PMC3372846
- DOI: 10.1111/j.1476-5381.2011.01696.x
Role of endothelin-1 in the hyper-responsiveness to nitrovasodilators following acute NOS inhibition
Abstract
Background and purpose: Acute NOS inhibition in humans and animals is associated with hypersensitivity to NO donors. The mechanisms underlying this phenomenon have not been fully elucidated. The purpose of the present study was to assess whether hypersensitivity to NOS-blockade is linked to endothelin-1 (ET-1) signalling.
Experimental approach: Sprague Dawley rats were instrumented with indwelling arterial and venous catheters for continuous assessments of haemodynamic parameters and drug delivery, respectively. Mesenteric arteries were isolated and tested for reactivity by wire myography.
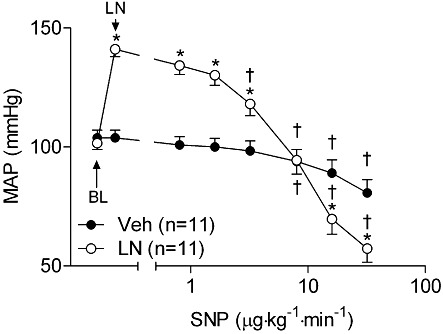
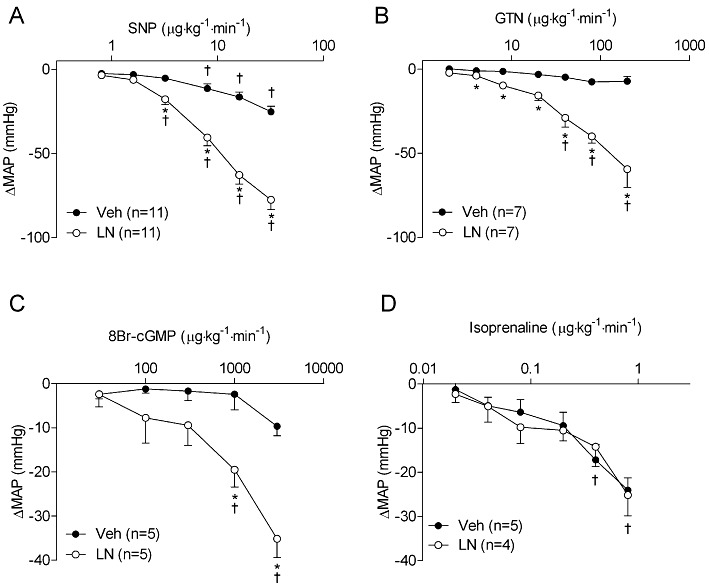
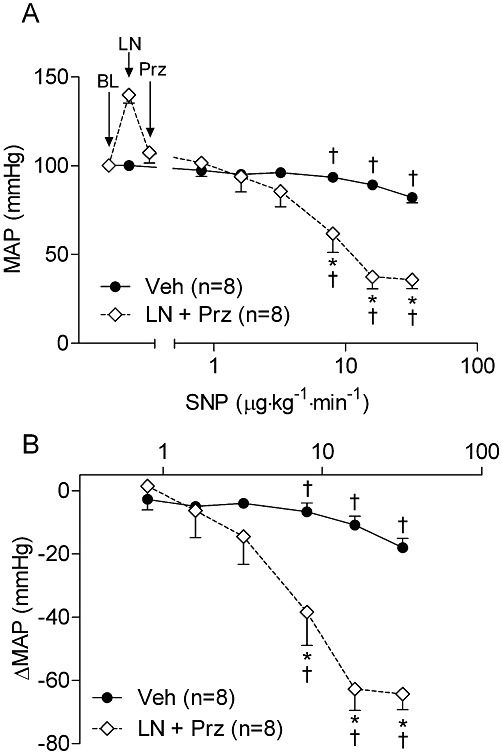
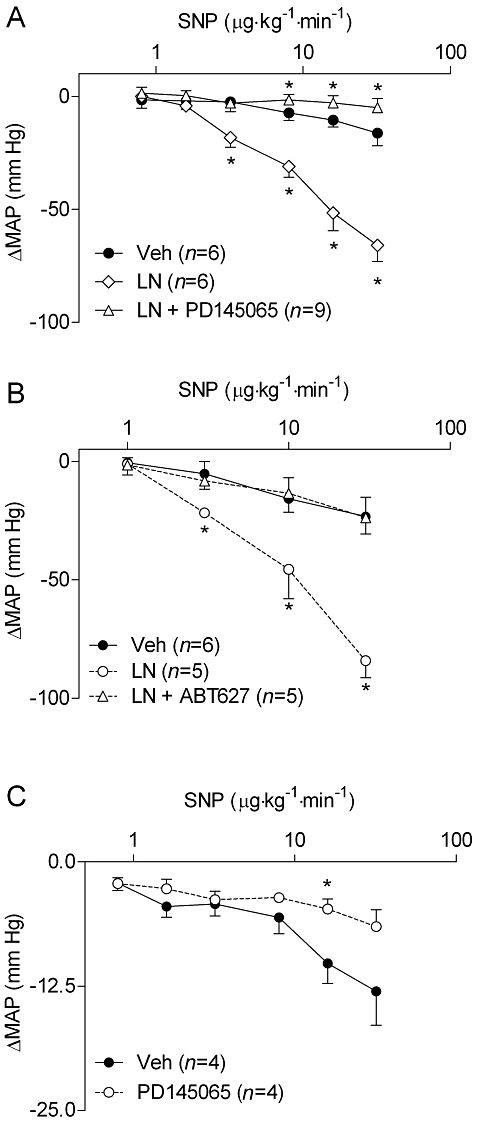
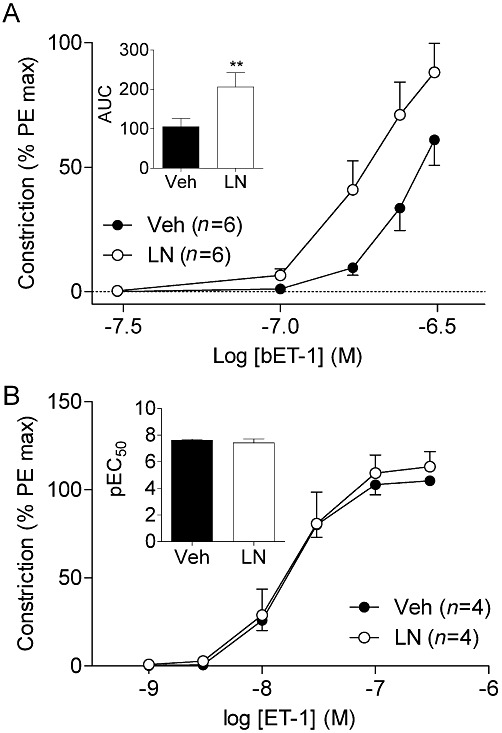
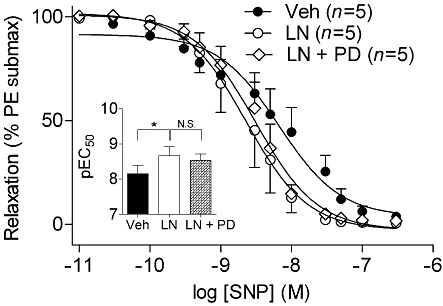
Key results: NOS blockade with L-N(G)-nitroarginine methyl ester (L-NAME) caused a pronounced increase in arterial blood pressure (BP) (∼40 mmHg). In L-NAME-treated animals, the dose of sodium nitroprusside (SNP) required to cause a significant reduction in arterial BP was lower than in vehicle-treated rats (P < 0.001), and the magnitude of the reduction in BP was greater. Similar results were obtained with other NO mimetics, but not isoprenaline; moreover, decreasing the BP back to baseline levels with prazosin after L-NAME treatment did not attenuate the hyper-responsiveness to NO donors. The increased responsiveness to NO donors was abolished by pretreatment with the ET(A/B) receptor antagonist, PD145065, or the ET(A) receptor-specific antagonist ABT627. Ex vivo, L-NAME treatment potentiated the constriction induced by big endothelin-1 (bET-1), the precursor to active ET-1, but had no effect on the ET-1-mediated constriction.
Conclusions and implications: These data suggest that the increased sensitivity to NO donors is mediated, at least in part, by ET-1 in vivo, and the mechanism may involve the conversion of bET-1 to ET-1.
© 2011 The Authors. British Journal of Pharmacology © 2011 The British Pharmacological Society.
Figures
References
-
- Bank N, Aynedjian HS, Khan GA. Mechanism of vasoconstriction induced by chronic inhibition of nitric oxide in rats. Hypertension. 1994;24:322–328. - PubMed
-
- Banting JD, Friberg P, Adams MA. Acute hypertension after nitric oxide synthase inhibition is mediated primarily by increased endothelin vasoconstriction. J Hypertens. 1996;14:975–981. - PubMed
-
- Banting JD, Thompson KE, Friberg P, Adams MA. Blunted cardiovascular growth induction during prolonged nitric oxide synthase blockade. Hypertension. 1997;30:416–421. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
