

Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method
- PMID: 21952777
- PMCID: PMC3396559
- DOI: 10.1007/s13361-011-0235-4
Many overlapping peptides for protein hydrogen exchange experiments by the fragment separation-mass spectrometry method
Abstract
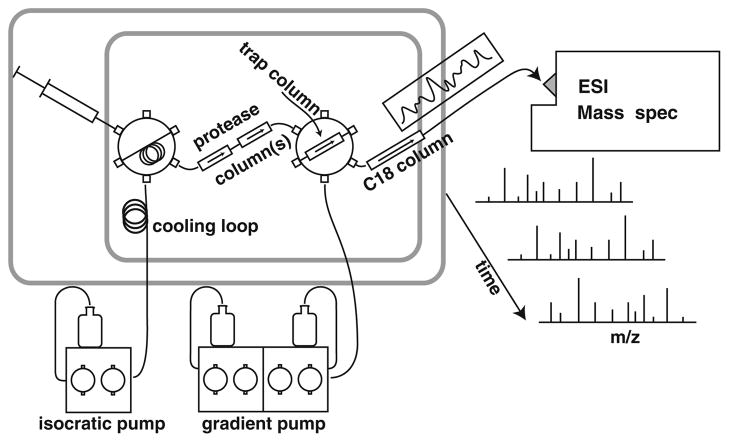
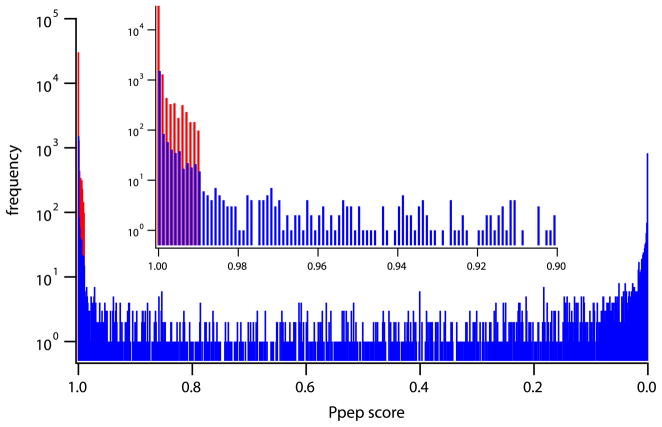
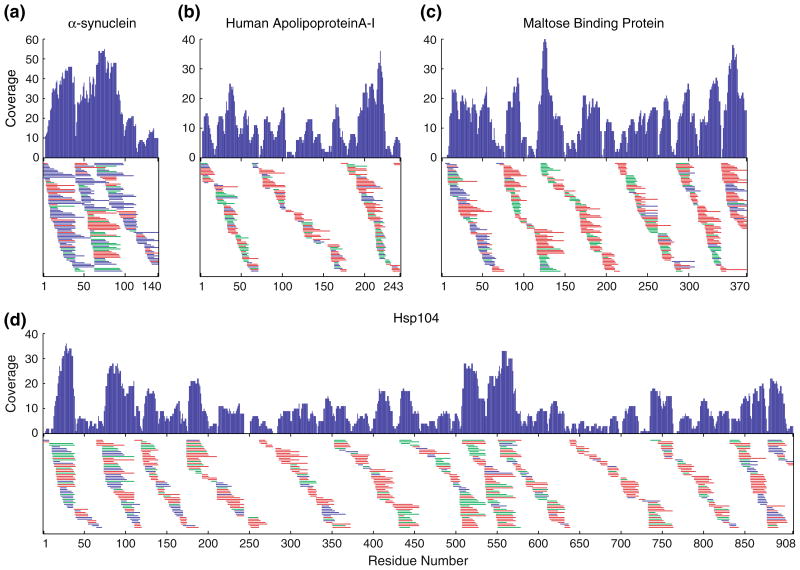
Measurement of the naturally occurring hydrogen exchange (HX) behavior of proteins can in principle provide highly resolved thermodynamic and kinetic information on protein structure, dynamics, and interactions. The HX fragment separation-mass spectrometry method (HX-MS) is able to measure hydrogen exchange in biologically important protein systems that are not accessible to NMR methods. In order to achieve high structural resolution in HX-MS experiments, it will be necessary to obtain many sequentially overlapping peptide fragments and be able to identify and analyze them efficiently and accurately by mass spectrometry. This paper describes operations which, when applied to four different proteins ranging in size from 140 to 908 residues, routinely provides hundreds of useful unique peptides, covering the entire protein length many times over. Coverage in terms of the average number of peptide fragments that span each amino acid exceeds 10. The ability to achieve these results required the integrated application of experimental methods that are described here and a computer analysis program, called ExMS, described in a following paper.
Figures
References
-
- Connelly GP, Bai Y, Jeng MF, Englander SW. Isotope Effects in Peptide Group Hydrogen Exchange. Proteins. 1993;17:87–92. - PubMed
-
- Molday RS, Englander SW, Kallen RG. Primary Structure Effects on Peptide Group Hydrogen Exchange. Biochemistry. 1972;11:150–158. - PubMed
-
- Hvidt A, Nielsen SO. Hydrogen Exchange in Proteins. Adv Protein Chem. 1966;21:287–386. - PubMed
-
- Linderstrøm-Lang K. Symposium on Protein Structure. Methuen; London: 1958. Deuterium Exchange and Protein structure.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
