

ExMS: data analysis for HX-MS experiments
- PMID: 21952778
- PMCID: PMC3398505
- DOI: 10.1007/s13361-011-0236-3
ExMS: data analysis for HX-MS experiments
Abstract
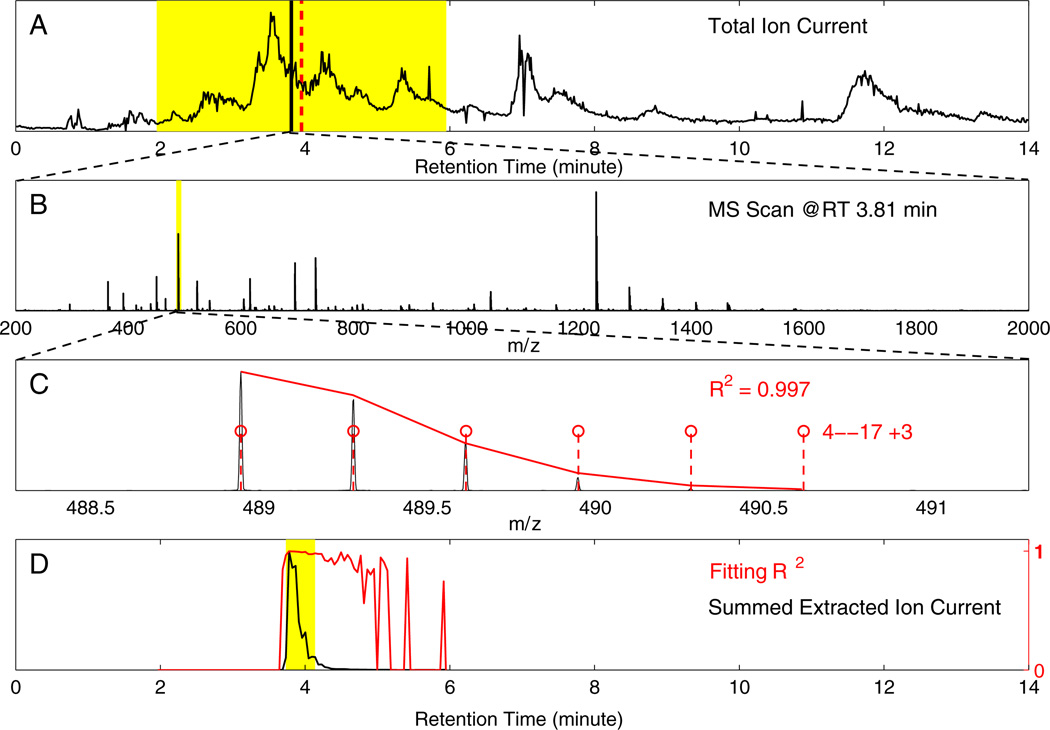
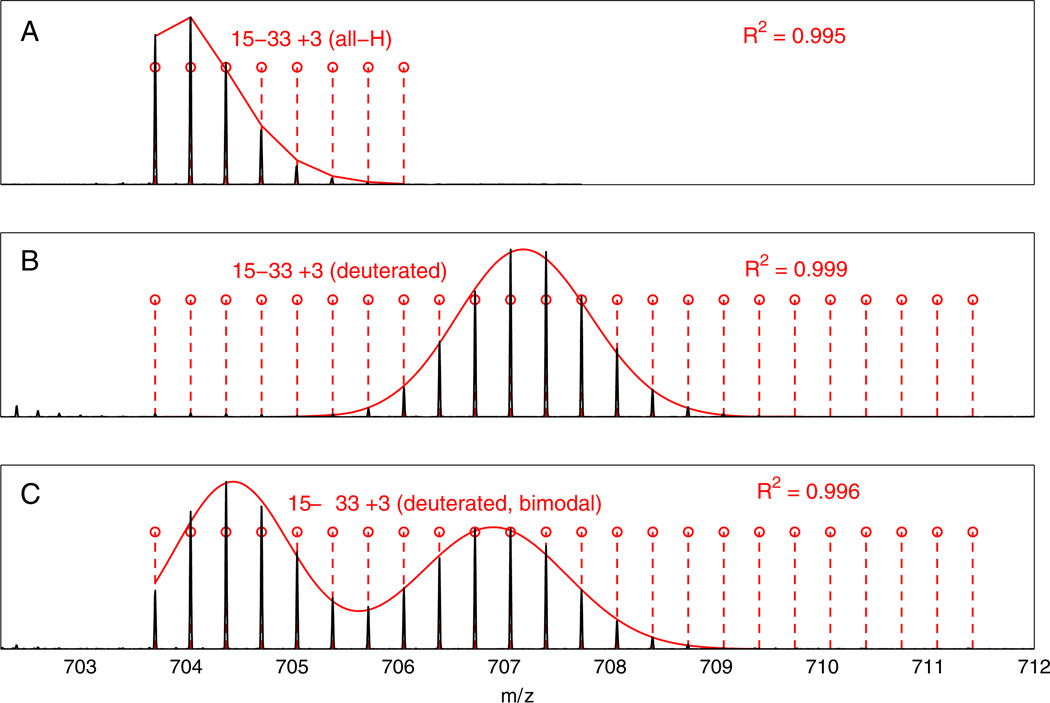
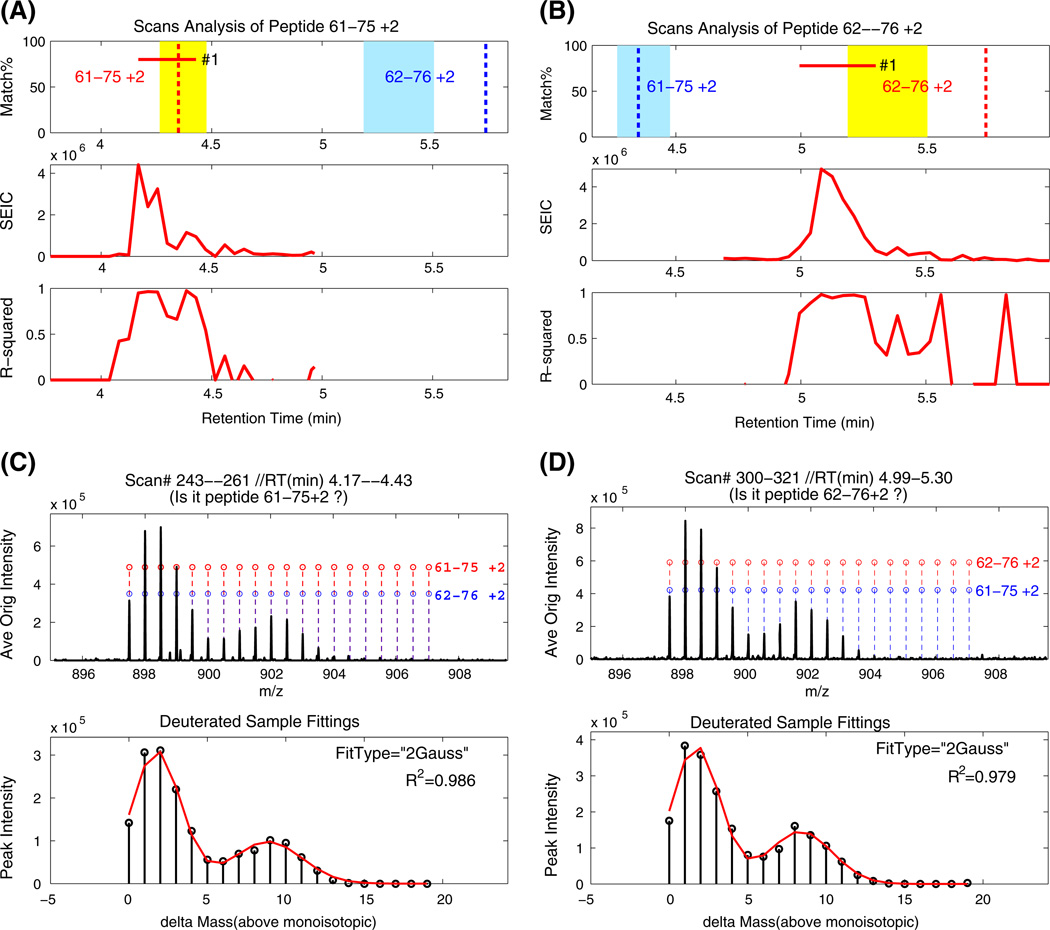
A previous paper considered the problems that presently limit the hydrogen exchange-mass spectrometry (HX-MS) method for studying the biophysical and functional properties of proteins. Many of these problems can be overcome by obtaining and analyzing hundreds of sequentially overlapping peptide fragments that cover the protein many times over (Mayne et al. J. Am. Soc. Mass Spectrom. 2011: 10.1007/s13361-011-0235-4). This paper describes a computer program called ExMS that furthers this advance by making it possible to efficiently process crowded mass spectra and definitively identify and characterize these many peptide fragments. ExMS automatically scans through high resolution MS data to find the individual isotopic peaks and isotopic envelopes of a list of peptides previously identified by MS/MS. It performs a number of tests to ensure correct identification in spite of peptide overlap in both chromatographic and mass spectrometric dimensions and possible multi-modal envelopes due to static or dynamic structural heterogeneity or HX EX1 behavior. The program can automatically process data from many sequential HX time points with no operator intervention at the rate of ~2 sec per peptide per HX time point using desktop computer equipment, but it also provides for rapid manual checking and decision when ambiguity exists. Additional subroutines can provide a step by step report of performance at each test along the way and parameter adjustment, deconvolute isotopic envelopes, and plot the time course of single and multi-modal H-D exchange. The program will be available on an open source basis at: http://HX2.med.upenn.edu/download.html.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
