

Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras
- PMID: 21952947
- PMCID: PMC4302750
- DOI: 10.1002/path.2971
Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras
Abstract
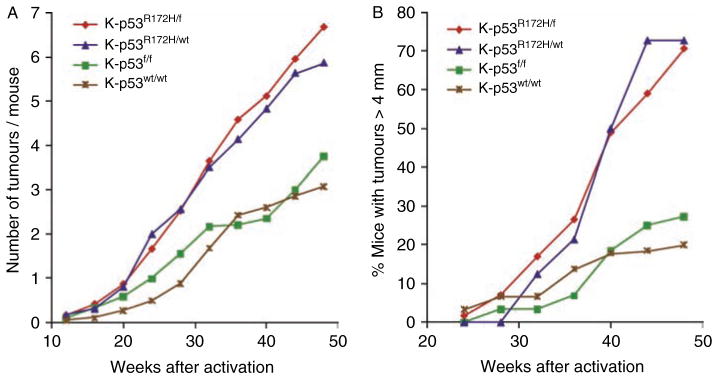
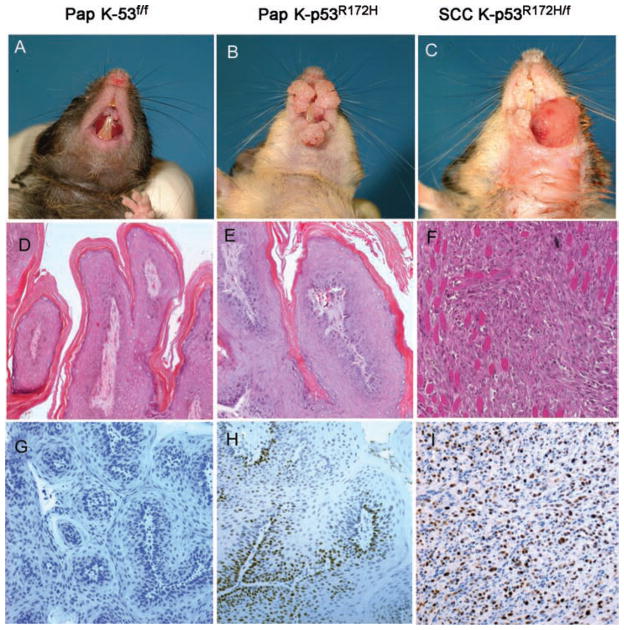
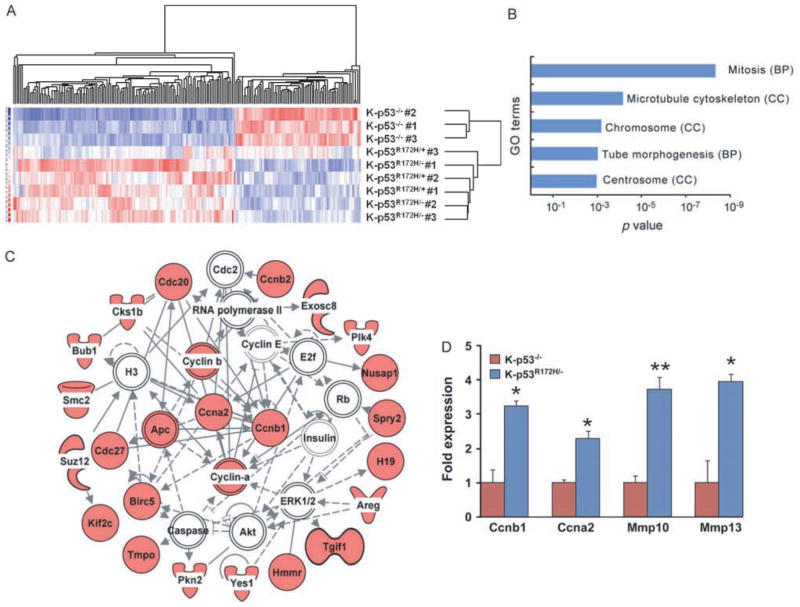
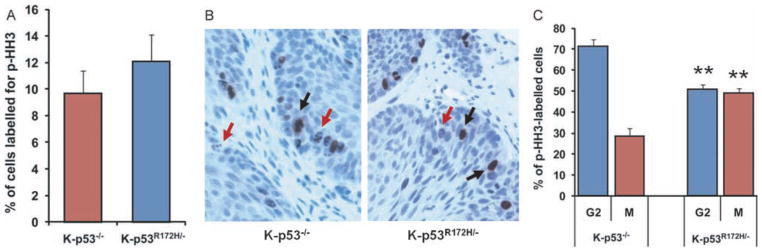
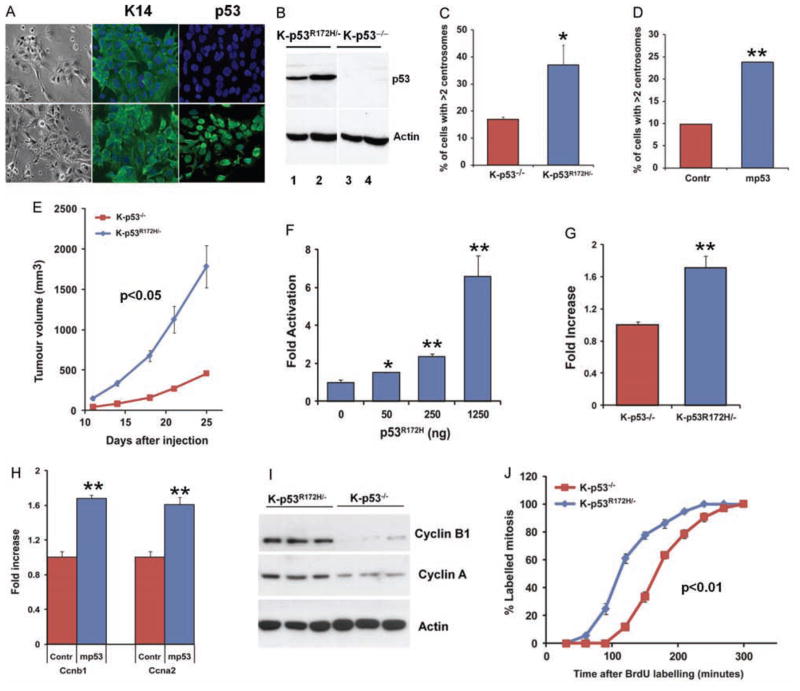
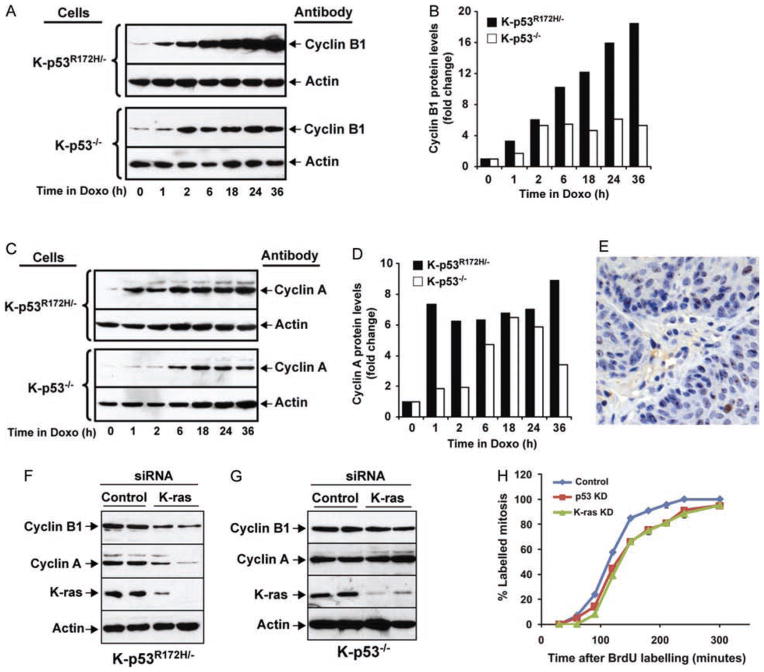
Mutations in p53 occur in over 50% of the human head and neck squamous cell carcinomas (SCCHN). The majority of these mutations result in the expression of mutant forms of p53, rather than deletions in the p53 gene. Some p53 mutants are associated with poor prognosis in SCCHN patients. However, the molecular mechanisms that determine the poor outcome of cancers carrying p53 mutations are unknown. Here, we generated a mouse model for SCCHN and found that activation of the endogenous p53 gain-of-function mutation p53$^{\rm{R172H}}$, but not deletion of p53, cooperates with oncogenic K-ras during SCCHN initiation, accelerates oral tumour growth, and promotes progression to carcinoma. Mechanistically, expression profiling of the tumours that developed in these mice and studies using cell lines derived from these tumours determined that mutant p53 induces the expression of genes involved in mitosis, including cyclin B1 and cyclin A, and accelerates entry in mitosis. Additionally, we discovered that this oncogenic function of mutant p53 was dependent on K-ras because the expression of cyclin B1 and cyclin A decreased, and entry in mitosis was delayed, after suppressing K-ras expression in oral tumour cells that express p53$^{\rm{R172H}}$. The presence of double-strand breaks in the tumours suggests that oncogene-dependent DNA damage resulting from K-ras activation promotes the oncogenic function of mutant p53. Accordingly, DNA damage induced by doxorubicin also induced increased expression of cyclin B1 and cyclin A in cells that express p53$^{\rm{R172H}}$. These findings represent strong in vivo evidence for an oncogenic function of endogenous p53 gain-of-function mutations in SCCHN and provide a mechanistic explanation for the genetic interaction between oncogenic K-ras and mutant p53.
Copyright © 2011 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Conflict of interest statement
No conflicts of interest were declared.
Figures
Comment in
-
Mutant p53 gain of function is interwoven into the hallmarks of cancer.J Pathol. 2011 Dec;225(4):475-8. doi: 10.1002/path.2988. J Pathol. 2011. PMID: 22025211
References
-
- Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. - PubMed
-
- Seiwert TY, Salama JK, Vokes EE. The chemoradiation paradigm in head and neck cancer. Nature Clin Pract Oncol. 2007;4:156–171. - PubMed
-
- Vokes EE, Weichselbaum RR, Lippman SM, et al. Head and neck cancer. N Engl J Med. 1993;328:184–194. - PubMed
-
- Culliney B, Birhan A, Young AV, et al. Management of locally advanced or unresectable head and neck cancer. Oncology (Williston Park) 2008;22:1152–1161. - PubMed
-
- Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–2492. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
