

Nuclear lamins and laminopathies
- PMID: 21953297
- PMCID: PMC6673656
- DOI: 10.1002/path.2999
Nuclear lamins and laminopathies
Abstract
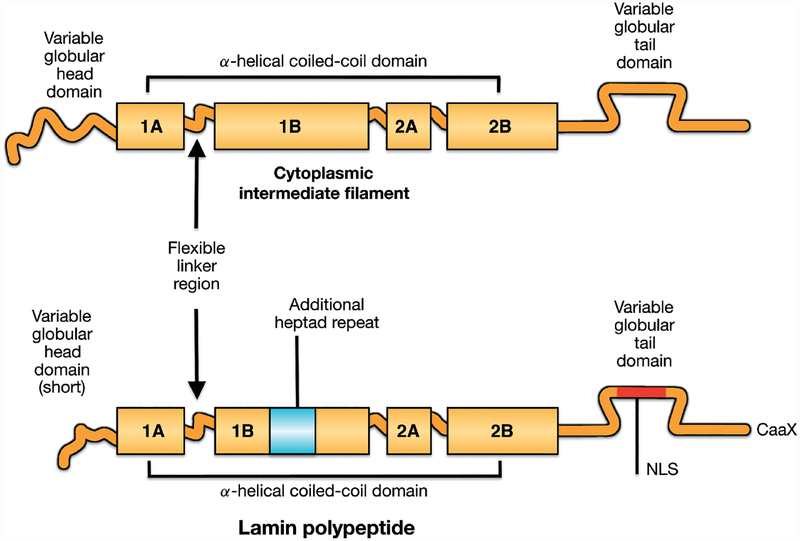
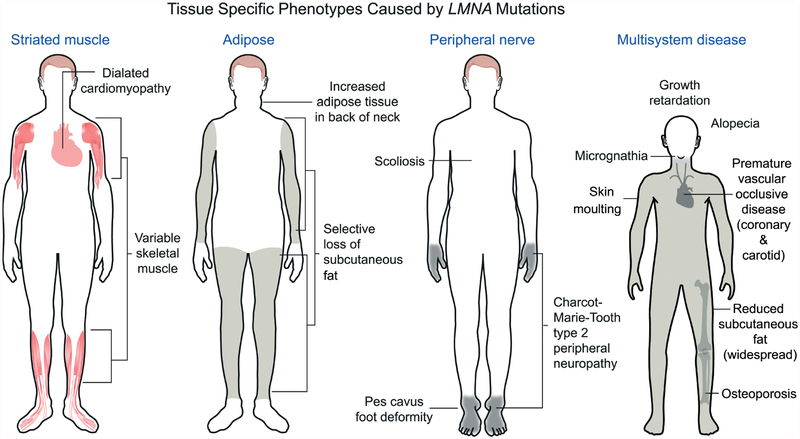
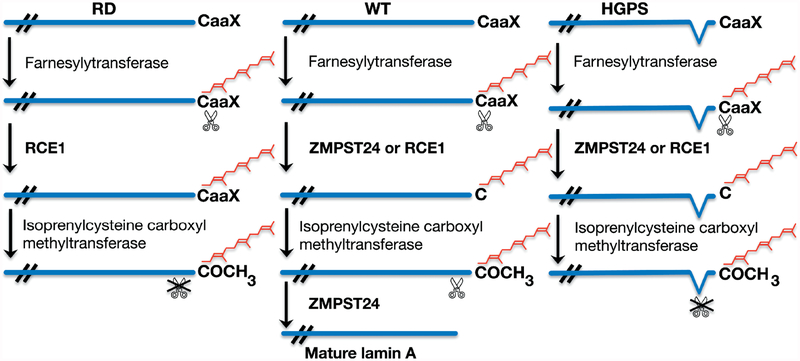
Nuclear lamins are intermediate filament proteins that polymerize to form the nuclear lamina on the inner aspect of the inner nuclear membrane. Long known to be essential for maintaining nuclear structure and disassembling/reassembling during mitosis in metazoans, research over the past dozen years has shown that mutations in genes encoding nuclear lamins, particularly LMNA encoding the A-type lamins, cause a broad range of diverse diseases, often referred to as laminopathies. Lamins are expressed in all mammalian somatic cells but mutations in their genes lead to relatively tissue-selective disease phenotypes in most cases. While mutations causing laminopathies have been shown to produce abnormalities in nuclear morphology, how these disease-causing mutations or resultant alterations in nuclear structure lead to pathology is only starting to be understood. Despite the incomplete understanding of pathogenic mechanisms underlying the laminopathies, basic research in cellular and small animal models has produced promising leads for treatments of these rare diseases.
Copyright © 2011 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Conflict of interest statement
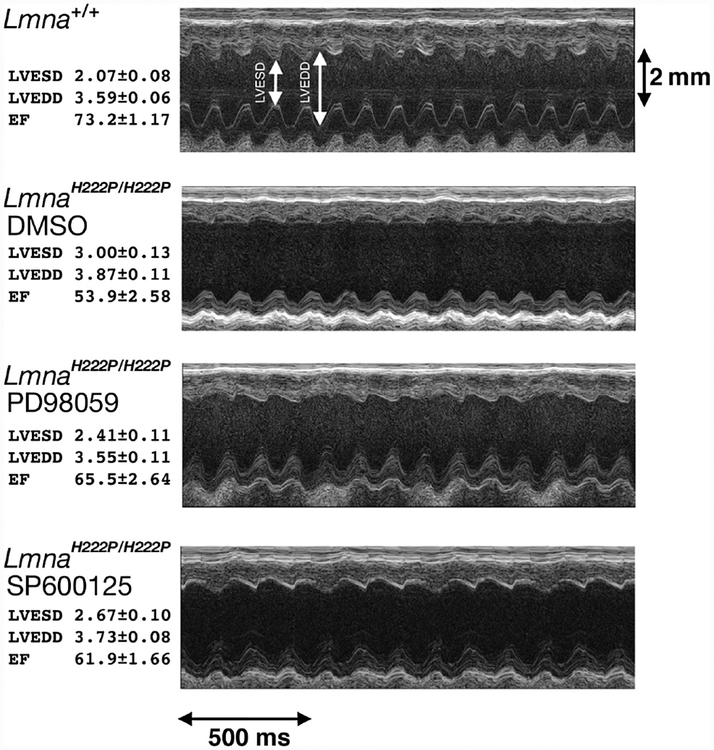
Conflict of interest: The author is an inventor on a pending PCT patent application on methods for treating and/or preventing cardiomyopathies by ERK and JNK inhibition, filed by the Trustees of Columbia University in the City of New York.
Figures
References
-
- Melcer S, Gruenbaum Y, Krohne G. Invertebrate lamins. Exp Cell Res 2007; 313: 2157–2166. - PubMed
-
- Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma? Nat Cell Biol 2004; 6: 1062–1067. - PubMed
-
- Frangioni J, Neel B Use of a general purpose mammalian expression vector for studying intracellular protein targeting: identification of critical residues in the nuclear lamin A/C nuclear localisation sequence. J Cell Sci 1993; 105: 481–488. - PubMed
-
- Dhe-Paganon S, Werner ED, Chi YI, et al. Structure of the globular tail of nuclear lamin. J Biol Chem 2002; 277: 17381–17384. - PubMed
-
- Krimm I, Östlund C, Gilquin B, et al. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002; 10: 811–823. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
