

Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils
- PMID: 21953712
- PMCID: PMC3377106
- DOI: 10.1002/emmm.201100176
Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils
Abstract
A decade has elapsed since the concept of oncogene addiction was first proposed. It postulates that - despite the diverse array of genetic lesions typical of cancer - some tumours rely on one single dominant oncogene for growth and survival, so that inhibition of this specific oncogene is sufficient to halt the neoplastic phenotype. A large amount of evidence has proven the pervasive power of this notion, both in basic research and in therapeutic applications. However, in the face of such a considerable body of knowledge, the intimate molecular mechanisms mediating this phenomenon remain elusive. At the clinical level, successful translation of the oncogene addiction model into the rational and effective design of targeted therapeutics against individual oncoproteins still faces major obstacles, mainly due to the emergence of escape mechanisms and drug resistance. Here, we offer an overview of the relevant literature, encompassing both biological aspects and recent clinical insights. We discuss the key advantages and pitfalls of this concept and reconsider it as an illustrative principle to guide post-genomic cancer research and drug development.
Copyright © 2011 EMBO Molecular Medicine.
Figures
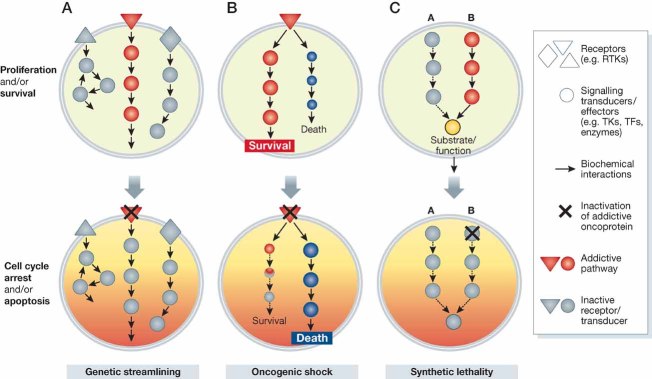
The ‘genetic streamlining’ theory postulates that non-essential pathways (top, light grey) are inactivated during tumour evolution, so that dominant, addictive pathways (red) are not surrogated by compensatory signals. Upon abrogation of dominant signals, there is a collapse in cellular fitness and cells experience cell-cycle arrest or apoptosis (bottom, red to yellow shading).
In the ‘oncogenic shock’ model, addictive oncoproteins (e.g. RTKs, red triangle) trigger at the same time pro-survival and pro-apoptotic signals (top, red and blue pathway, respectively). Under normal conditions, the pro-survival outputs dominate over the pro-apoptotic ones (top), but following blockade of the addictive receptor, the rapid decline in the activity of survival pathways (dashed lines, bottom) subverts this balance in favour of death-inducing signals, which tend to last longer and eventually lead to apoptotic death.
Two genes are considered to be in a synthetic lethal relationship when loss of one or the other is still compatible with survival but loss of both is fatal. In the top panel, biochemical inactivation of pathway A (grey) has no effect on cell viability because pathway B (red), which converges at some point on a common substrate or effector (yellow), has compensating activity. When the integrity of pathway B is disrupted (bottom), the common downstream biochemical function is lost and again cancer cells may experience cell cycle arrest or apoptosis.
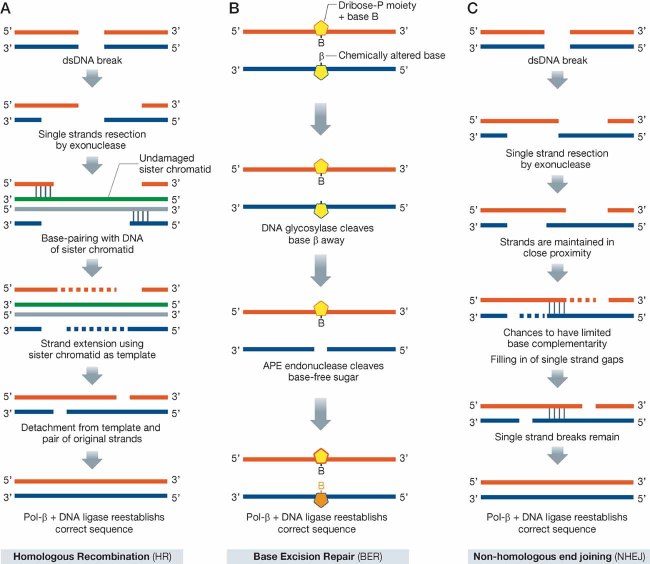
HR safeguards genome integrity in late S/G2 phases of the cell cycle and relies on the ability to use the recently formed sister chromatids as a template to guide repair of the damaged strands. Exonuclease activity produces two single-stranded (ss) DNA ends, one at each side of a double-strand (ds) DNA break (top). Each of the two ssDNA ends pair with complementary sequences of the sister chromatid (unwinding of sister chromatids is not represented here for simplicity); elongation is then performed by DNA polymerase. Subsequent release from the sister chromatid, pairing of the elongated ssDNA ends, further elongation and, ultimately, ligation give rise to the wild-type sequence. BRCA1 and BRCA2, among many other proteins, are part of the HR machinery.
BER senses chemically altered bases (β) with a minimal effect on double helix topology. Glycosylases first cleave the bond linking the base with the deoxyribose (middle top); this is in turn excised by an apurinic/apyrimidinic endonuclease (APE) with high affinity for base-free sugars (middle bottom). DNA polymerase β replaces the missing nucleotide, which is finally ligated to reconstitute the correct sequence.
When homologous sequences are not available as templates, the ssDNA ends generated by exonucleases can be joined together by a small number of base pairs. This event if followed by filling of the gaps in each strand and ligation of any remaining ssDNA breaks. Of note, the sequence repaired by this NHEJ repair system lacks some of the bases originally present in the undamaged DNA and is therefore intrinsically error-prone.
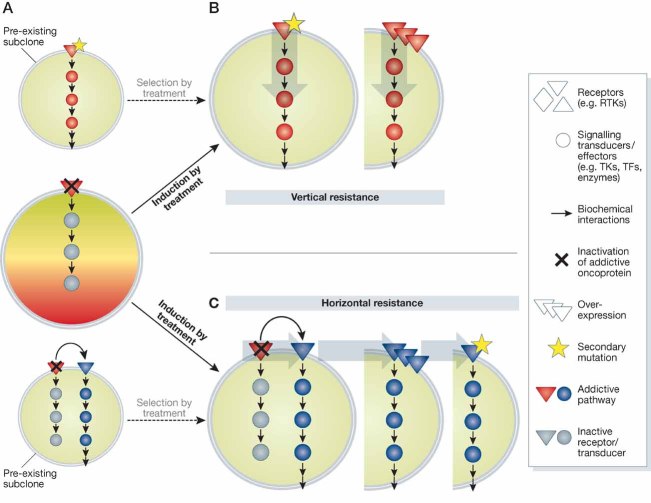
A. When the addictive oncoprotein is inhibited, viability is seriously compromised and cancer cells stop proliferating or die (middle). Addiction to a certain oncogene might not be a homogeneous characteristic of the cancer cell population, and drug-insensitive subclones (top and bottom) may co-exist with sensitive cells.
B, C. Fuelled by genomic instability, targeted treatment of addicted cells may result in induction (arrows, solid line) of secondary resistance through either mutation (star) or amplification/overexpression of relevant signalling nodes. This may happen in a vertical fashion, by alterations involving downstream effectors of the original addictive pathway (B), or in a horizontal fashion, when a parallel signalling axis surrogates target blockade (C). If resistant clones are already present at the beginning of treatment (panel A), these may be selected by drug exposure (arrows, dashed line) until they outcompete sensitive cells, resulting in ‘acquired’ resistance as well.
References
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
