

SILACtor: software to enable dynamic SILAC studies
- PMID: 21954881
- PMCID: PMC3255570
- DOI: 10.1021/ac2017053
SILACtor: software to enable dynamic SILAC studies
Abstract
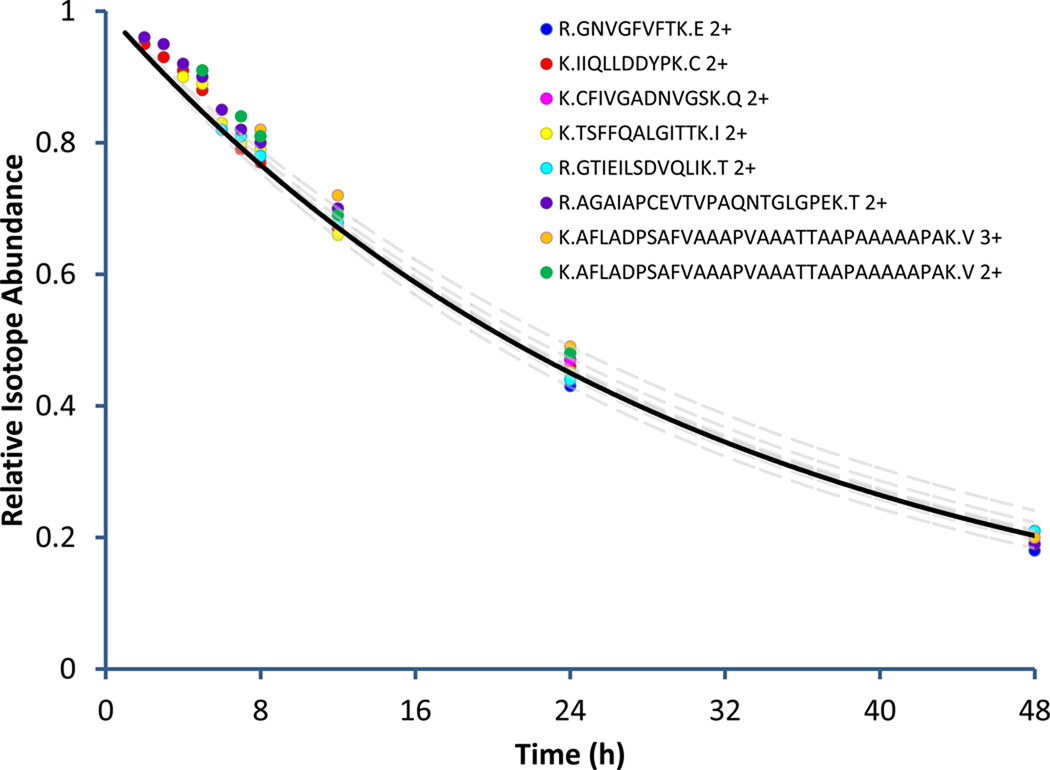
Stable isotope labeling by amino acids in cell culture (SILAC) is a versatile tool in proteomics that has been used to explore protein turnover on a large scale. However, these studies pose a significant undertaking that can be greatly simplified through the use of computational tools that automate the data analysis. While SILAC technology has enjoyed rapid adoption through the availability of several software tools, algorithms do not exist for the automated analysis of protein turnover data generated using SILAC technology. Presented here is a software tool, SILACtor, designed to trace and compare SILAC-labeled peptides across multiple time points. SILACtor is used to profile protein turnover rates for more than 500 HeLa cell proteins using a SILAC label-chase approach. Additionally, SILACtor contains a method for the automated generation of accurate mass and retention time inclusion lists that target peptides of interest showing fast or slow turnover rates relative to the other peptides observed in the samples. SILACtor enables improved protein turnover studies using SILAC technology and also provides a framework for features extensible to comparative SILAC analyses and targeted methods.
Figures




References
-
- Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–386. - PubMed
-
- Ong SE, Foster LJ, Mann M. Methods. 2003;29:124–130. - PubMed
-
- Cox J, Mann M. Nat Biotechnol. 2008;26:1367–1372. - PubMed
-
- Li XJ, Zhang H, Ranish JA, Aebersold R. Anal Chem. 2003;75:6648–6657. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
