

Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome
- PMID: 21957152
- PMCID: PMC3205568
- DOI: 10.1101/gr.116509.110
Late-replicating heterochromatin is characterized by decreased cytosine methylation in the human genome
Abstract
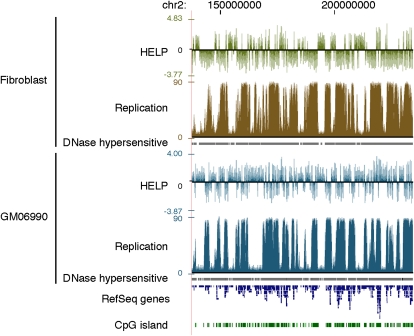
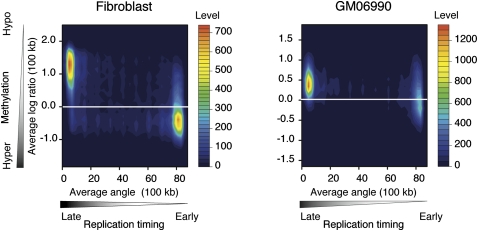
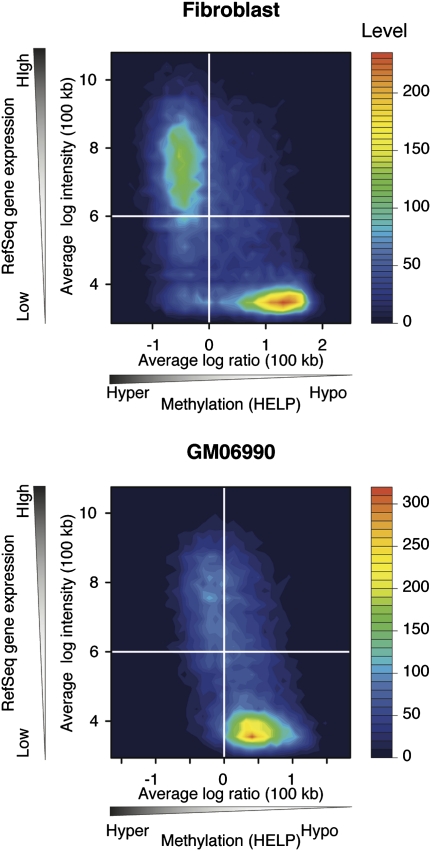
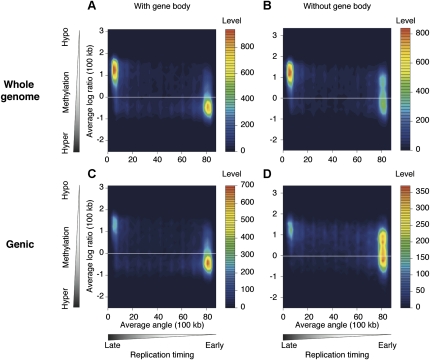
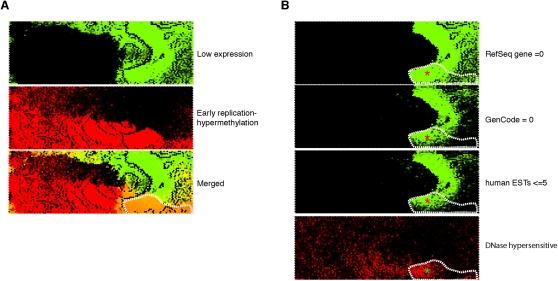
Heterochromatin is believed to be associated with increased levels of cytosine methylation. With the recent availability of genome-wide, high-resolution molecular data reflecting chromatin organization and methylation, such relationships can be explored systematically. As well-defined surrogates for heterochromatin, we tested the relationship between DNA replication timing and DNase hypersensitivity with cytosine methylation in two human cell types, unexpectedly finding the later-replicating, more heterochromatic regions to be less methylated than early replicating regions. When we integrated gene-expression data into the study, we found that regions of increased gene expression were earlier replicating, as previously identified, and that transcription-targeted cytosine methylation in gene bodies contributes to the positive correlation with early replication. A self-organizing map (SOM) approach was able to identify genomic regions with early replication and increased methylation, but lacking annotated transcripts, loci missed in simple two variable analyses, possibly encoding unrecognized intergenic transcripts. We conclude that the relationship of cytosine methylation with heterochromatin is not simple and depends on whether the genomic context is tandemly repetitive sequences often found near centromeres, which are known to be heterochromatic and methylated, or the remaining majority of the genome, where cytosine methylation is targeted preferentially to the transcriptionally active, euchromatic compartment of the genome.
Figures

References
-
- Aran D, Toperoff G, Rosenberg M, Hellman A 2011. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genet 20: 670–680 - PubMed
-
- Backdahl L, Herberth M, Wilson G, Tate P, Campos LS, Cortese R, Eckhardt F, Beck S 2009. Gene body methylation of the dimethylarginine dimethylamino-hydrolase 2 (Ddah2) gene is an epigenetic biomarker for neural stem cell differentiation. Epigenetics 4: 248–254 - PubMed
-
- Berezney R, Dubey DD, Huberman JA 2000. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 108: 471–484 - PubMed
-
- Bernardino-Sgherri J, Flagiello D, Dutrillaux B 2002. Overall DNA methylation and chromatin structure of normal and abnormal X chromosomes. Cytogenet Genome Res 99: 85–91 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials