

A comparative analysis of exome capture
- PMID: 21958622
- PMCID: PMC3308060
- DOI: 10.1186/gb-2011-12-9-r97
A comparative analysis of exome capture
Abstract
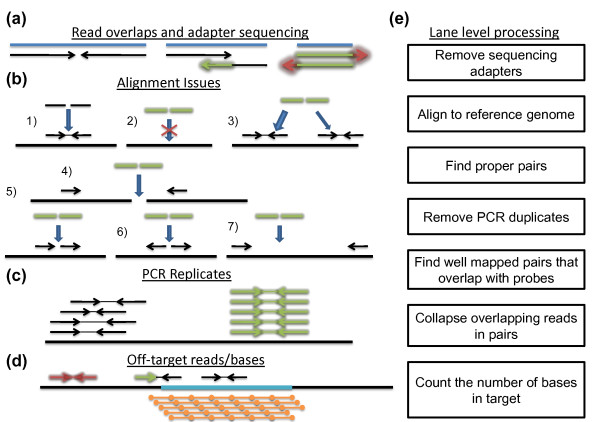
Background: Human exome resequencing using commercial target capture kits has been and is being used for sequencing large numbers of individuals to search for variants associated with various human diseases. We rigorously evaluated the capabilities of two solution exome capture kits. These analyses help clarify the strengths and limitations of those data as well as systematically identify variables that should be considered in the use of those data.
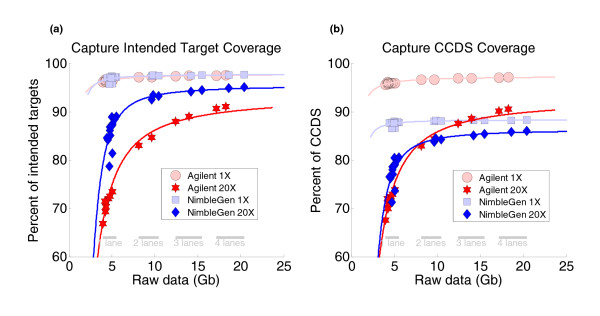
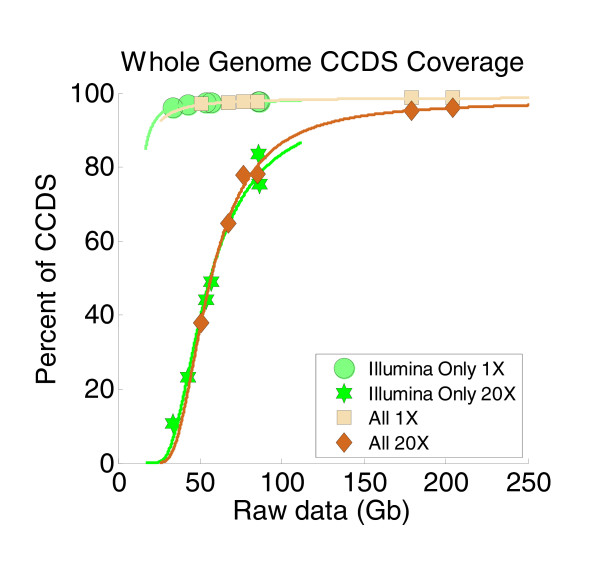
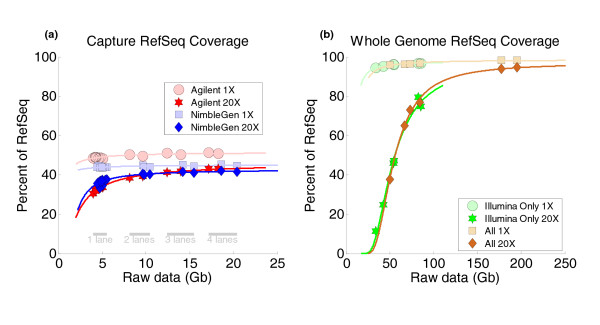
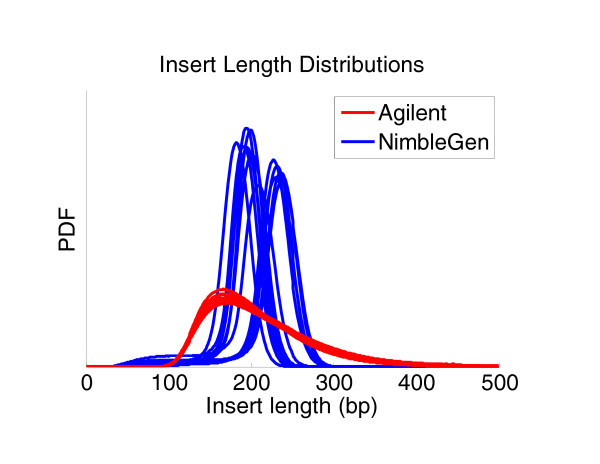
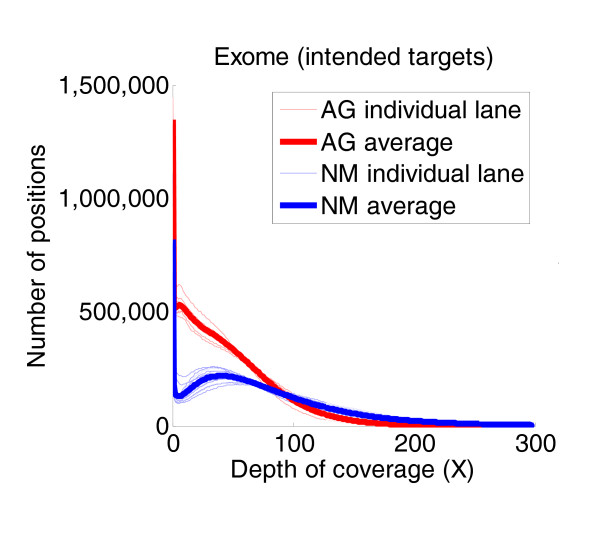
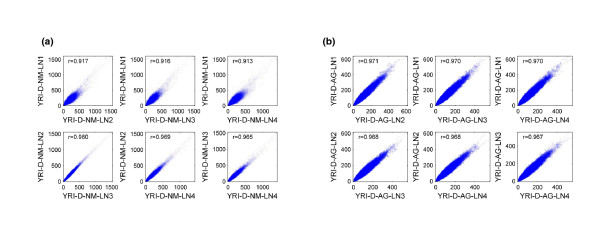
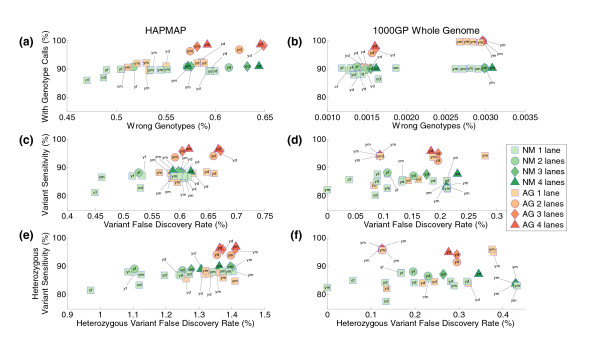
Results: Each exome kit performed well at capturing the targets they were designed to capture, which mainly corresponds to the consensus coding sequences (CCDS) annotations of the human genome. In addition, based on their respective targets, each capture kit coupled with high coverage Illumina sequencing produced highly accurate nucleotide calls. However, other databases, such as the Reference Sequence collection (RefSeq), define the exome more broadly, and so not surprisingly, the exome kits did not capture these additional regions.
Conclusions: Commercial exome capture kits provide a very efficient way to sequence select areas of the genome at very high accuracy. Here we provide the data to help guide critical analyses of sequencing data derived from these products.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
