

Accurate identification of A-to-I RNA editing in human by transcriptome sequencing
- PMID: 21960545
- PMCID: PMC3246201
- DOI: 10.1101/gr.124107.111
Accurate identification of A-to-I RNA editing in human by transcriptome sequencing
Abstract
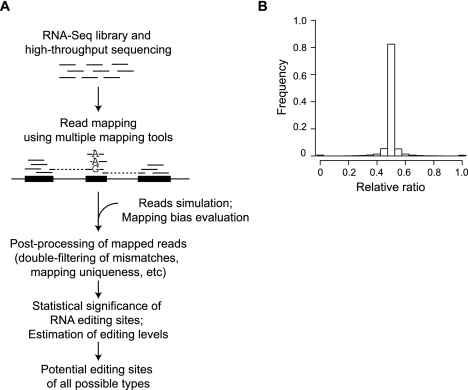
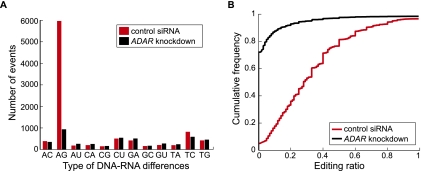
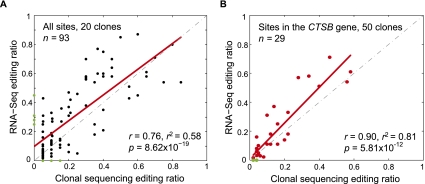
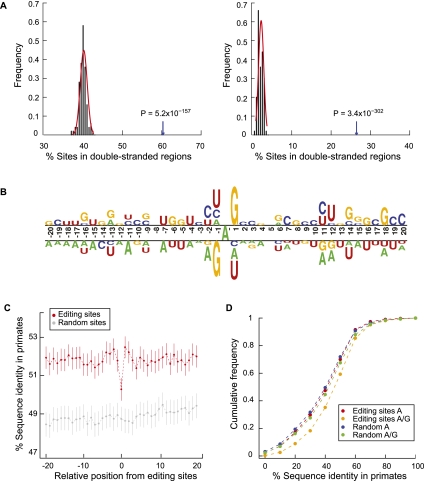
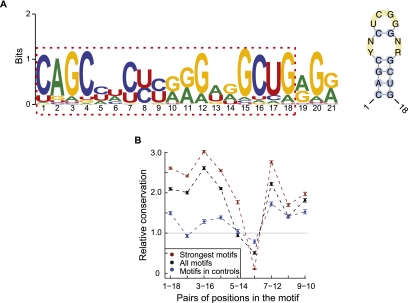
RNA editing enhances the diversity of gene products at the post-transcriptional level. Approaches for genome-wide identification of RNA editing face two main challenges: separating true editing sites from false discoveries and accurate estimation of editing levels. We developed an approach to analyze transcriptome sequencing data (RNA-seq) for global identification of RNA editing in cells for which whole-genome sequencing data are available. We applied the method to analyze RNA-seq data of a human glioblastoma cell line, U87MG. Around 10,000 DNA-RNA differences were identified, the majority being putative A-to-I editing sites. These predicted A-to-I events were associated with a low false-discovery rate (∼5%). Moreover, the estimated editing levels from RNA-seq correlated well with those based on traditional clonal sequencing. Our results further facilitated unbiased characterization of the sequence and evolutionary features flanking predicted A-to-I editing sites and discovery of a conserved RNA structural motif that may be functionally relevant to editing. Genes with predicted A-to-I editing were significantly enriched with those known to be involved in cancer, supporting the potential importance of cancer-specific RNA editing. A similar profile of DNA-RNA differences as in U87MG was predicted for another RNA-seq data set obtained from primary breast cancer samples. Remarkably, significant overlap exists between the putative editing sites of the two transcriptomes despite their difference in cell type, cancer type, and genomic backgrounds. Our approach enabled de novo identification of the RNA editome, which sets the stage for further mechanistic studies of this important step of post-transcriptional regulation.
Figures
References
-
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases