

Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology
- PMID: 21960726
- PMCID: PMC3299091
- DOI: 10.1161/CIRCRESAHA.111.243154
Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology
Retraction in
-
Retraction of: Role of Cardiac Stem Cells in Cardiac Pathophysiology: A Paradigm Shift in Human Myocardial Biology.Circ Res. 2019 Feb 15;124(4):e24. doi: 10.1161/RES.0000000000000248. Circ Res. 2019. PMID: 30582464 Free PMC article. No abstract available.
Expression of concern in
-
Expression of Concern.Circ Res. 2019 Jan 18;124(2):e4-e5. doi: 10.1161/RES.0000000000000241. Circ Res. 2019. PMID: 30582460 No abstract available.
-
Expression of Concern.Circulation. 2019 Jan 15;139(3):e5-e6. doi: 10.1161/CIR.0000000000000639. Circulation. 2019. PMID: 30615475 No abstract available.
Abstract
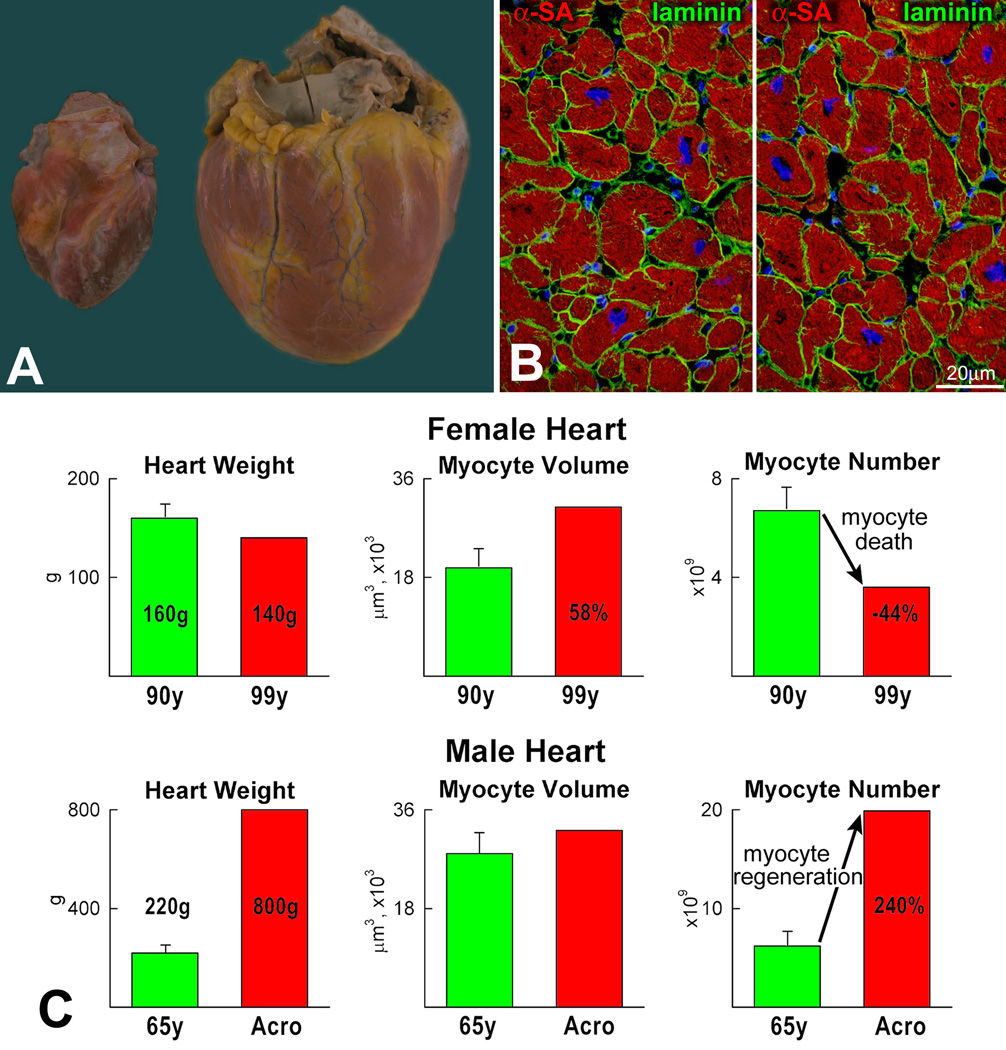
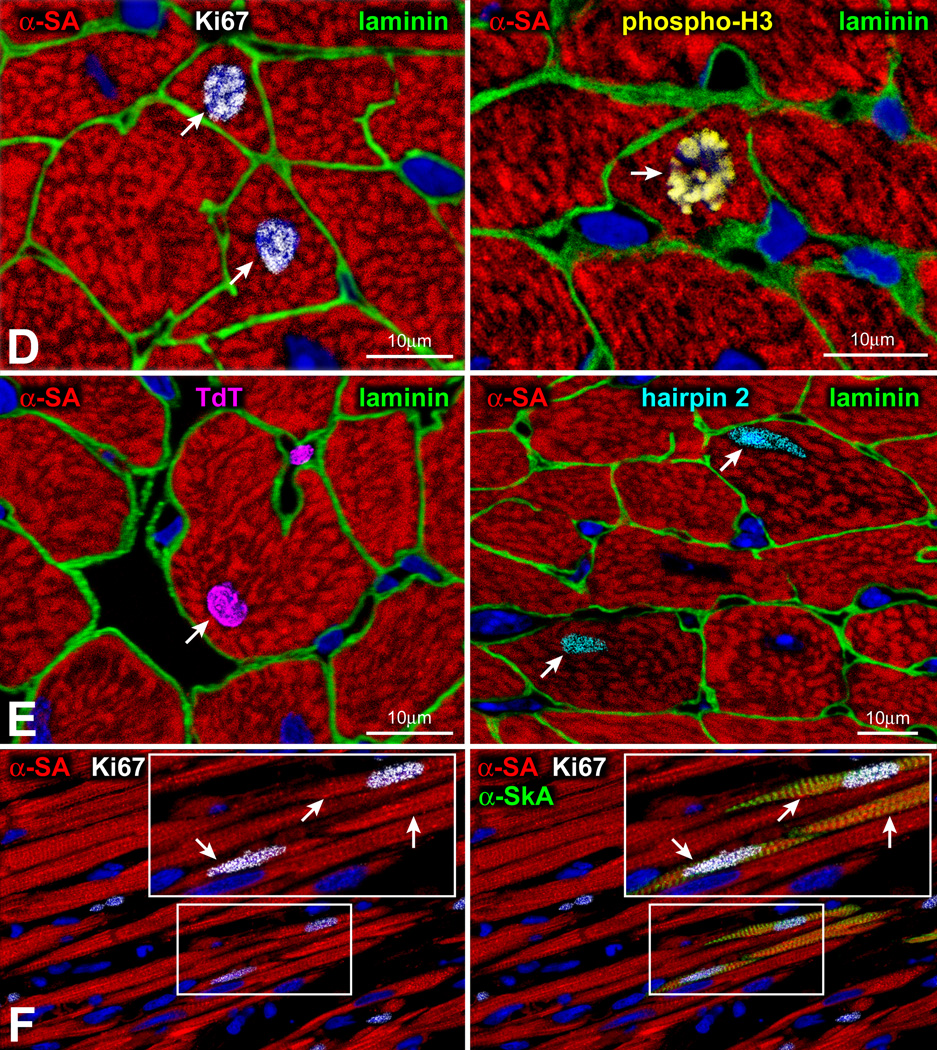
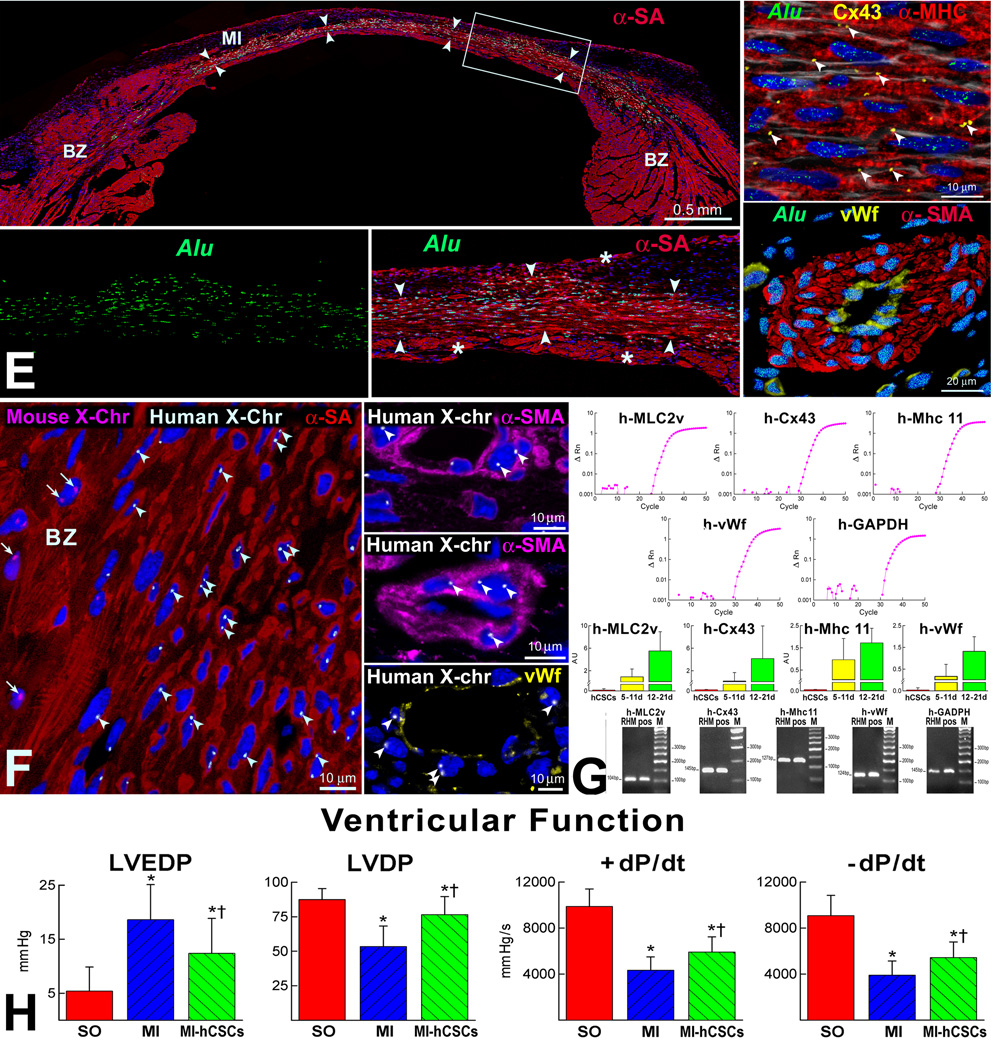
For nearly a century, the human heart has been viewed as a terminally differentiated postmitotic organ in which the number of cardiomyocytes is established at birth, and these cells persist throughout the lifespan of the organ and organism. However, the discovery that cardiac stem cells live in the heart and differentiate into the various cardiac cell lineages has changed profoundly our understanding of myocardial biology. Cardiac stem cells regulate myocyte turnover and condition myocardial recovery after injury. This novel information imposes a reconsideration of the mechanisms involved in myocardial aging and the progression of cardiac hypertrophy to heart failure. Similarly, the processes implicated in the adaptation of the infarcted heart have to be dissected in terms of the critical role that cardiac stem cells and myocyte regeneration play in the restoration of myocardial mass and ventricular function. Several categories of cardiac progenitors have been described but, thus far, the c-kit-positive cell is the only class of resident cells with the biological and functional properties of tissue specific adult stem cells.
Figures













Comment in
-
Cardiomyocyte renewal in humans.Circ Res. 2012 Jan 6;110(1):e17-8; author reply e19-21. doi: 10.1161/CIRCRESAHA.111.259598. Circ Res. 2012. PMID: 22223215 No abstract available.
References
-
- Anversa P, Olivetti G. The Heart. New York: Oxford University Press; 2002. Cellular basis of physiological and pathological myocardial growth. Handbook of physiology: the cardiovascular system; pp. 75–144.
-
- Hill JA, Olson EN. Mechanisms of disease cardiac plasticity. N Engl J Med. 2008;358:1370–1380. - PubMed
-
- Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. - PubMed
-
- Hill JA. Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc Med. 2003;13:316–322. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 AG017042/AG/NIA NIH HHS/United States
- R01 HL111183/HL/NHLBI NIH HHS/United States
- R01 HL075480/HL/NHLBI NIH HHS/United States
- R01 HL114346/HL/NHLBI NIH HHS/United States
- P01 HL092868/HL/NHLBI NIH HHS/United States
- R01 AG037490/AG/NIA NIH HHS/United States
- R01 HL039902/HL/NHLBI NIH HHS/United States
- R01 HL105532/HL/NHLBI NIH HHS/United States
- R01 AG037495/AG/NIA NIH HHS/United States
- R01 AG026107/AG/NIA NIH HHS/United States
- R01 HL065577/HL/NHLBI NIH HHS/United States
- R01 HL065573/HL/NHLBI NIH HHS/United States
- R37 HL081737/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
