

doi: 10.1016/j.cell.2011.09.008.
Clan genomics and the complex architecture of human disease
Affiliations
- PMID: 21962505
- PMCID: PMC3656718
- DOI: 10.1016/j.cell.2011.09.008
Item in Clipboard
Clan genomics and the complex architecture of human disease
Cell.
.
Abstract
Human diseases are caused by alleles that encompass the full range of variant types, from single-nucleotide changes to copy-number variants, and these variations span a broad frequency spectrum, from the very rare to the common. The picture emerging from analysis of whole-genome sequences, the 1000 Genomes Project pilot studies, and targeted genomic sequencing derived from very large sample sizes reveals an abundance of rare and private variants. One implication of this realization is that recent mutation may have a greater influence on disease susceptibility or protection than is conferred by variations that arose in distant ancestors.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
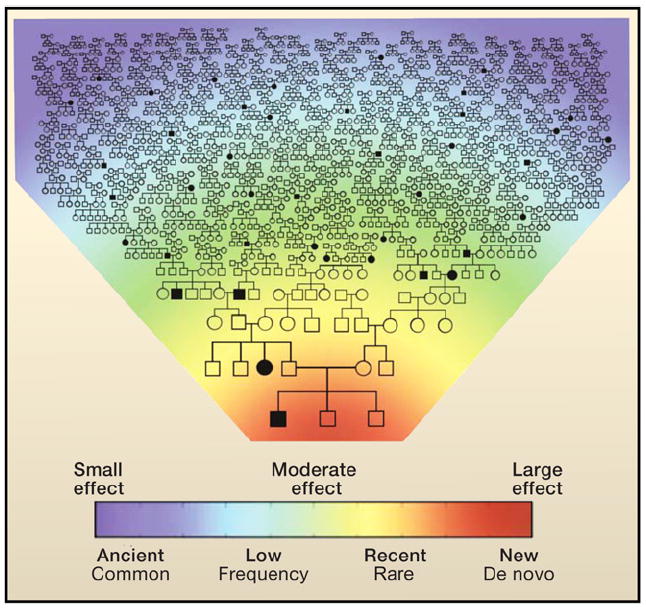
Heat map and extended pedigree showing the conceptual relationship among de novo mutations leading to disease (red), recent mutations with moderate effects arising within a clan (yellow and green), and older common variants with small effects segregating in the population (blue). An individual’s genetic disease risk emerges from the collection of variants he or she has inherited from both parental lineages of distant ancestors (typically common and of individually small effect), more recent ancestors (rare, but potentially larger effect), and de novo mutations.

This figure demonstrates “clan genomics,” wherein the combinations of alleles one inherits from his or her nearest relatives profoundly affect clinical outcome. In these illustrative pedigrees, different combinations of ABCA4 alleles can affect age-of-onset of Stargardt macular dystrophy (STGD; MIM 601691) (A), Mendelian versus multifactorial trait (i.e., Stargardt disease versus age-related macular degeneration [AMD]) (B), or retinal disease type (i.e., Stargardt disease versus retinitis pigmentosa [RP; MIM 601718]) (C). (D) Differing SH3TC2 alleles result in recessive Charcot-Marie-Tooth disease (CMT; MIM 601596), dominant axonal neuropathy, or the complex trait of carpal tunnel syndrome.
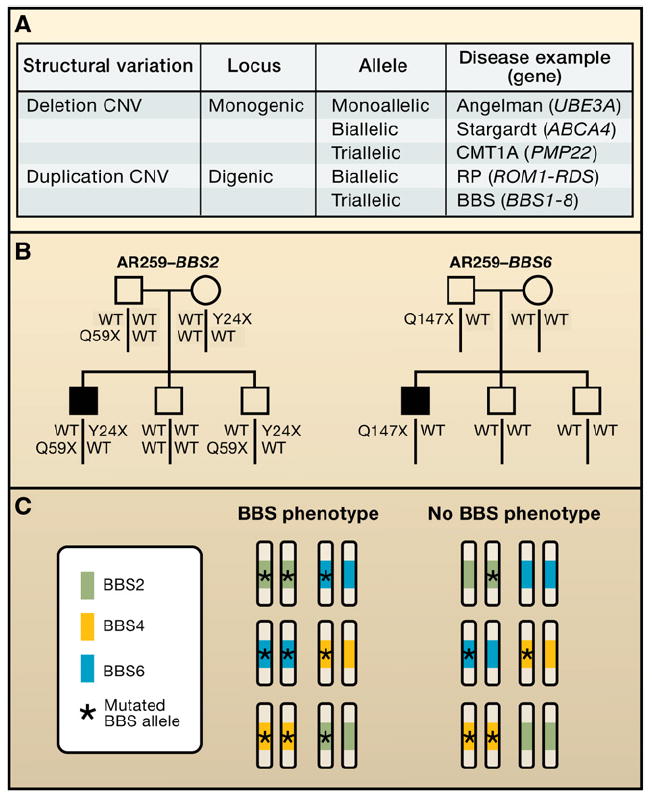
(A) In classical Mendelian disease, for a recessive, monogenic disease, at that single locus there is biallelic inheritance (highlighted in box). Examples could be either Stargardt macular dystrophy or cystic fibrosis, which are both due to point mutations in ATP-binding cassette (ABC) transporter genes. However, at some loci in the human genome, imprinting results in monoallelic expression, and the disease phenotype will occur in a manner dependent on the parent of origin of the specific mutation, either by deletion copy number variants (CNV) or uniparental disomy (UPD). The example given is the Angelman syndrome with point mutations in the UBE3A gene. The CMT1A locus (17p12) represents a triallelic locus whereby because of the duplication, there are three copies of the PMP22 gene. None of the copies have point mutations in them, but it takes three copies to convey the clinical phenotype. Other examples of disease allele transmission include interactions between two or potentially more genes. In the classic model of digenic inheritance, the phenotype of retinitis pigmentosa has been shown to be due to heterozygous point mutations in the ROM1 gene in combination with heterozygous point mutations at the RDS locus. Thus there is biallelic digenic inheritance. Note that a genomic deletion CNV renders a locus monoallelic, whereas a duplication CNV results in a triallelic locus. (B) Bardet-Biedl syndrome (BBS), traditionally thought of as a recessive trait, can sometimes result from three mutant alleles, two of which come from one locus, and one from another locus. This is an example of digenic triallelic inheritance. (C) A single pedigree illustrates triallelic inheritance for BBS. Standard pedigree symbols are used; filled squares, affected with BBS. Alleles segregating at two distinct loci (BBS2 and BBS6) are shown, one in each pedigree. WT, wild-type or normal allele.
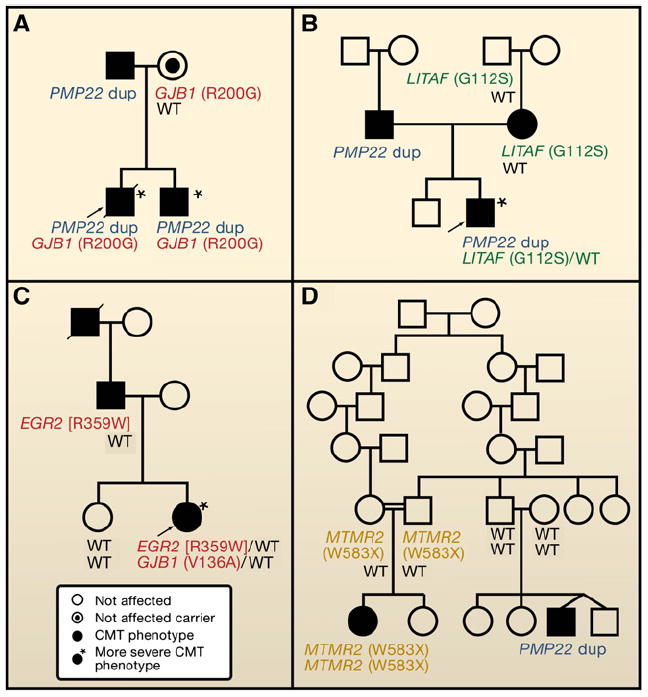
Pedigrees of families segregating Charcot-Marie-Tooth (CMT) neuropathy, illustrating that disease severity is directly related to pathogenic mutational burden. (A–C) Mutations at two different CMT loci result in a more severe phenotype. These double heterozygotes may be due to either a single-nucleotide variant (SNV) + copy-number variant (CNV) (A and B) or two SNV (C) (Chung et al., 2005; Hodapp et al., 2006; Meggouh et al., 2005). (D) In a single family, disease results from homozygous MTMR2 mutation (likely related to consanguinity) or de novo CNV—the CMT1A duplication (PMP22) (Verny et al., 2004); an example of clan genomics.
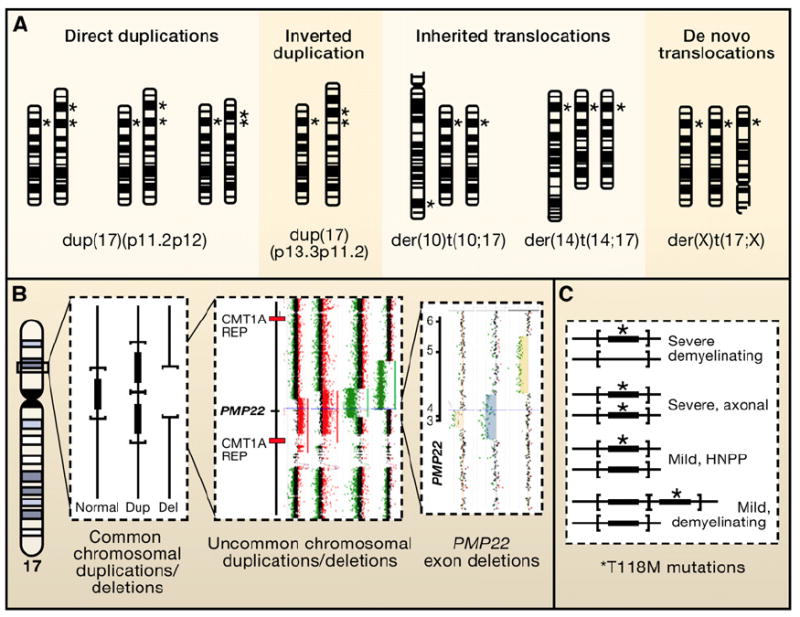
(A) Chromosomal duplication mapping wherein chromosomally visible duplication abnormalities, as evidenced by altered G-banding patterns, are used to delineate the portion of the genome responsible for the reduced motor nerve conduction velocities that accompany the demyelinating form of Charcot-Marie-Tooth disease (CMT1A; MIM 118220). Several different chromosomal abnormalities have been reported in association with a CMT1 phenotype. Note, different chromosome 17 abnormalities including direct duplications, inverted duplications, and inherited as well as de novo translocations have been reported with complex phenotypes that include CMT. If the duplicated genomic interval encompasses the 17p12 dark G-band where the PMP22 gene maps (*), then the patient will have a demyelinating neuropathy, as evidenced by decreased motor nerve conduction velocities, as part of their clinical phenotype. (B) Submicroscopic genomic rearrangements associated with neuropathy. Vertical lines represent a “blow-up” of the genomic interval within 17p12 containing the PMP22 gene (filled rectangle). The horizontal parentheses delimit the rearranged interval for the common deletion (depicted by absence of vertical line) and duplication (two copies of gene and interval). To the right are rare-sized copy-number variants (CNV) depicting genomic deletion (green dots on array CGH) versus duplication (red dots on array). (C) shows genotype/phenotype correlations between PMP22 point mutations associated with neuropathy. The T118M missense amino acid substitution in PMP22 appears to be a reduced penetrance loss-of-function allele. As a heterozygous mutation it can result in a mild hereditary neuropathy with liability to pressure palsies (HNPP) phenotype in some individuals; as a homozygous allele it can convey a severe axonal neuropathy. Interestingly, when the T118M allele occurs in combination with the HNPP deletion, a severe demyelinating phenotype results. Of further interest, when the T118M allele occurs in combination with the CMT1A duplication, the loss-of-function missense amino substitution appears to mitigate some of the consequences of the gain-of-function duplication CNV.
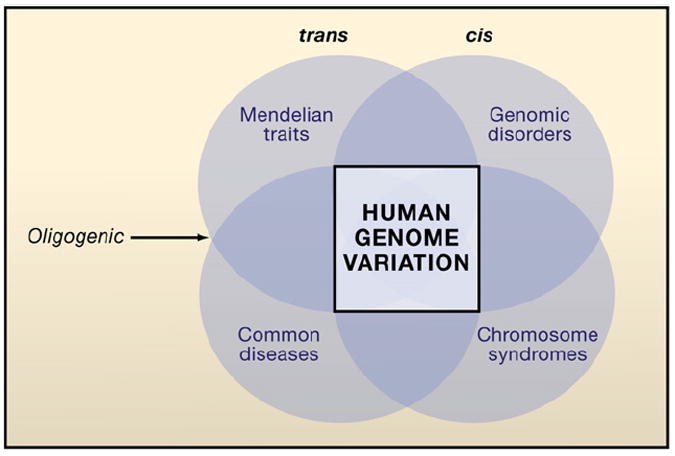
The square (center) represents genomic variation that can influence the different categories of genetic disease. The circles represent the overlapping categories of human disease with darker regions depicting intersection with greater overlap in the underlying genetic influences on these given disease categories. A unified model for human genetic disease proposes that all major categories of disease with genetic influence—Mendelian disease, common disease or complex traits, genomic disorders, and chromosomal syndromes—can be explained by variation in DNA sequence (SNV) or copy number (CNV) from a “wild-type” diploid state. Whereas trans-genetic interactions at a single locus (alleles) or between loci may contribute to Mendelian disease and complex traits, cis-genetic interactions can be important to phenotypic manifestations in genomic disorders (CNV) and chromosomal syndromes (segmental aneuploidy). Digenic and triallelic inheritance bridge Mendelian traits and complex disease; each represents an oligogenic inheritance model.
References
-
- Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. - PubMed
-
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997a;277:1805–1807. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
