

Modeling human disease in humans: the ciliopathies
- PMID: 21962508
- PMCID: PMC3202432
- DOI: 10.1016/j.cell.2011.09.014
Modeling human disease in humans: the ciliopathies
Abstract
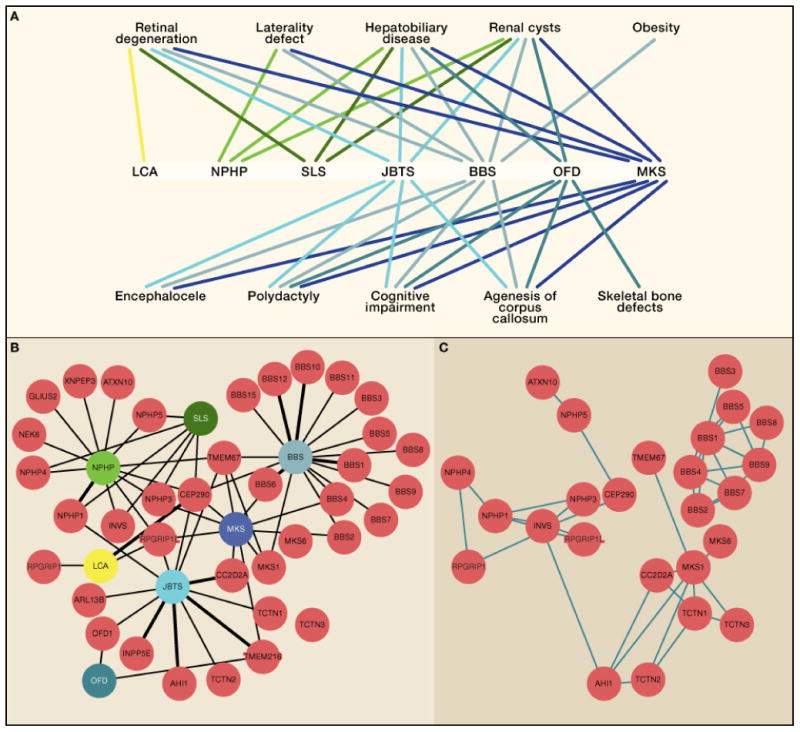
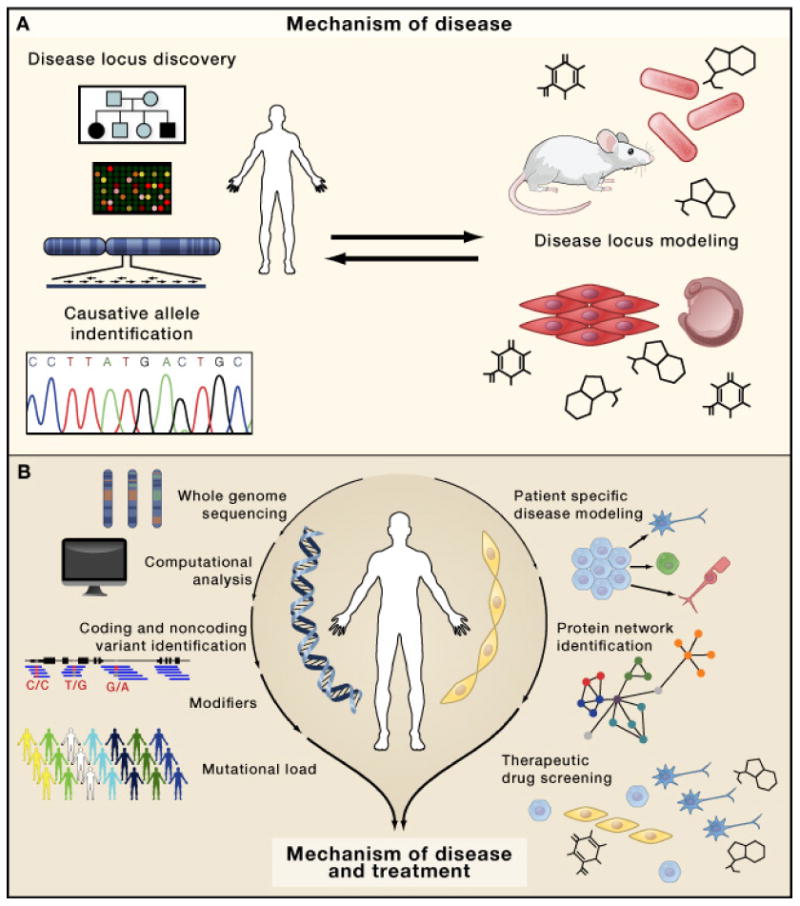
Soon, the genetic basis of most human Mendelian diseases will be solved. The next challenge will be to leverage this information to uncover basic mechanisms of disease and develop new therapies. To understand how this transformation is already beginning to unfold, we focus on the ciliopathies, a class of multi-organ diseases caused by disruption of the primary cilium. Through a convergence of data involving mutant gene discovery, proteomics, and cell biology, more than a dozen phenotypically distinguishable conditions are now united as ciliopathies. Sitting at the interface between simple and complex genetic conditions, these diseases provide clues to the future direction of human genetics.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures

References
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. - PubMed
-
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
