

Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting
- PMID: 21964591
- PMCID: PMC5417176
- DOI: 10.1210/me.2011-1124
Myostatin induces degradation of sarcomeric proteins through a Smad3 signaling mechanism during skeletal muscle wasting
Erratum in
-
Erratum.Mol Endocrinol. 2015 Jan;29(1):153. doi: 10.1210/me.2014-1357. Mol Endocrinol. 2015. PMID: 25549158 Free PMC article. No abstract available.
Retraction in
-
Retraction.Mol Endocrinol. 2016 Feb;30(2):274. doi: 10.1210/me.2015-1318. Epub 2016 Jan 8. Mol Endocrinol. 2016. PMID: 26745670 Free PMC article. No abstract available.
Abstract
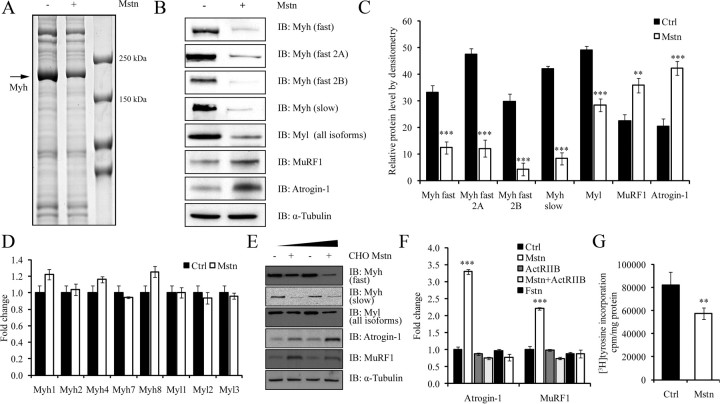
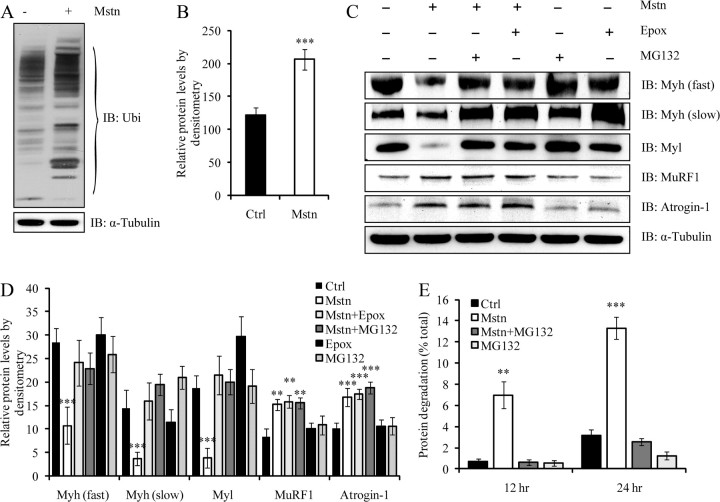
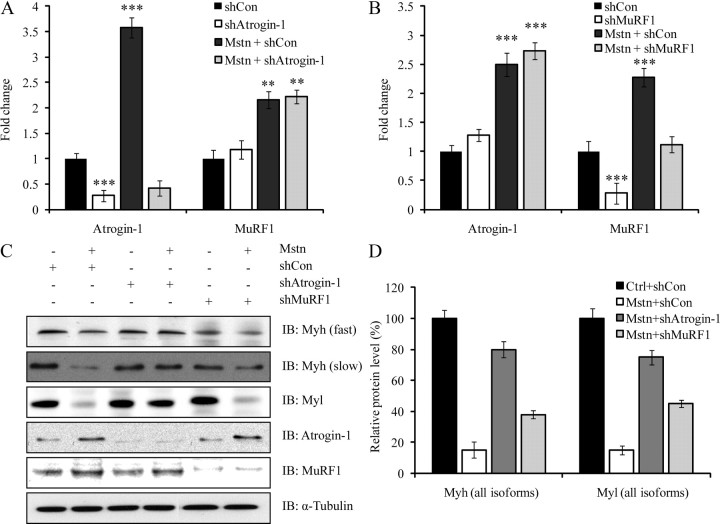
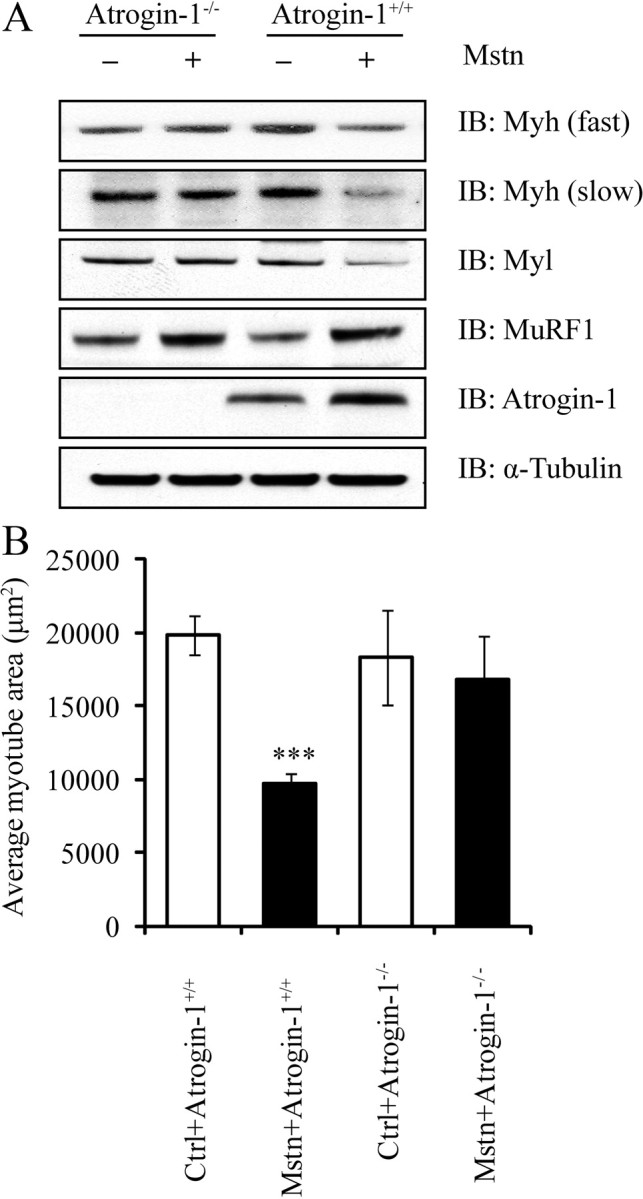
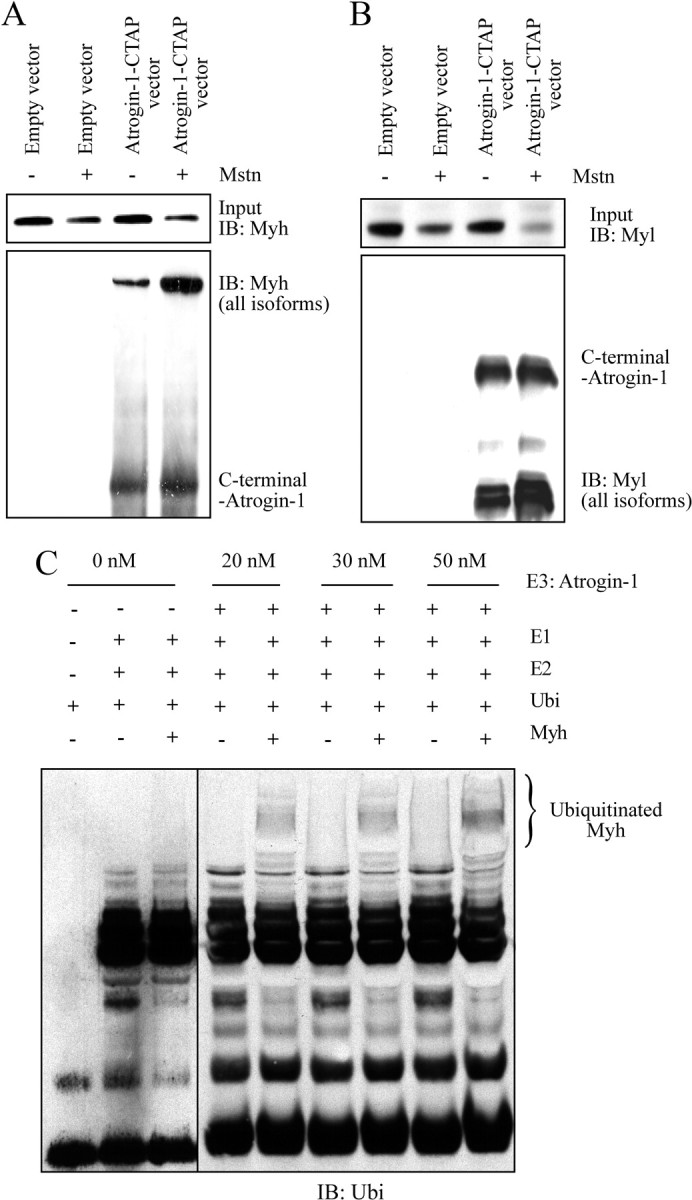
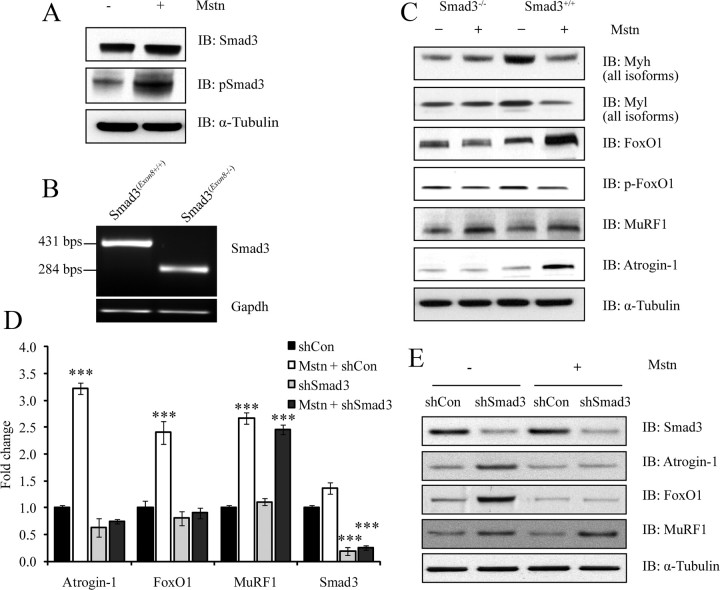
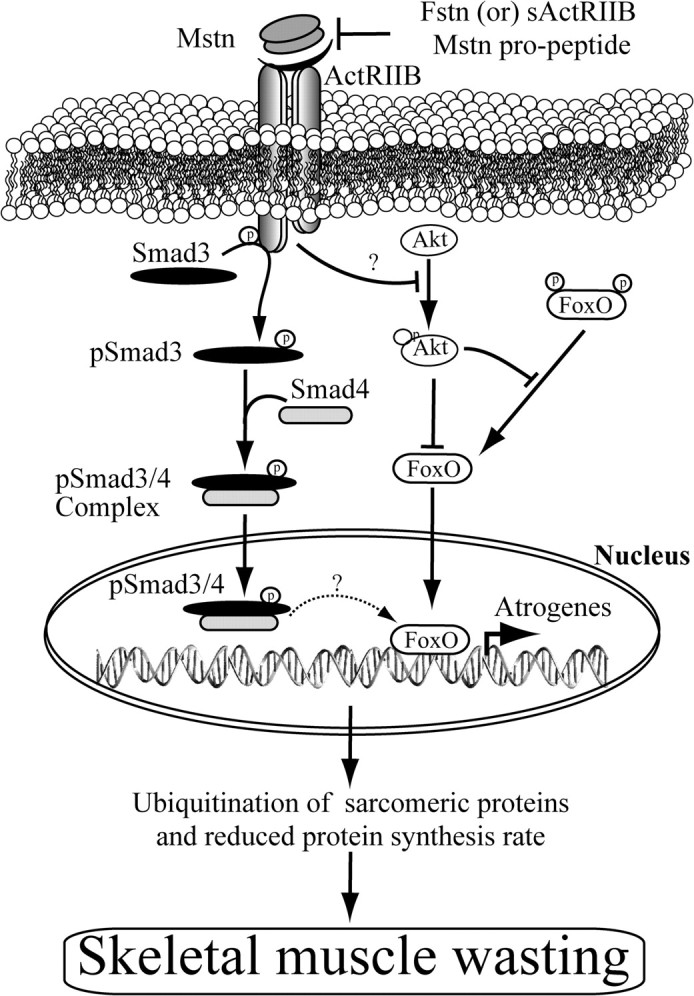
Ubiquitination-mediated proteolysis is a hallmark of skeletal muscle wasting manifested in response to negative growth factors, including myostatin. Thus, the characterization of signaling mechanisms that induce the ubiquitination of intracellular and sarcomeric proteins during skeletal muscle wasting is of great importance. We have recently characterized myostatin as a potent negative regulator of myogenesis and further demonstrated that elevated levels of myostatin in circulation results in the up-regulation of the muscle-specific E3 ligases, Atrogin-1 and muscle ring finger protein 1 (MuRF1). However, the exact signaling mechanisms by which myostatin regulates the expression of Atrogin-1 and MuRF1, as well as the proteins targeted for degradation in response to excess myostatin, remain to be elucidated. In this report, we have demonstrated that myostatin signals through Smad3 (mothers against decapentaplegic homolog 3) to activate forkhead box O1 and Atrogin-1 expression, which further promotes the ubiquitination and subsequent proteasome-mediated degradation of critical sarcomeric proteins. Smad3 signaling was dispensable for myostatin-dependent overexpression of MuRF1. Although down-regulation of Atrogin-1 expression rescued approximately 80% of sarcomeric protein loss induced by myostatin, only about 20% rescue was seen when MuRF1 was silenced, implicating that Atrogin-1 is the predominant E3 ligase through which myostatin manifests skeletal muscle wasting. Furthermore, we have highlighted that Atrogin-1 not only associates with myosin heavy and light chain, but it also ubiquitinates these sarcomeric proteins. Based on presented data we propose a model whereby myostatin induces skeletal muscle wasting through targeting sarcomeric proteins via Smad3-mediated up-regulation of Atrogin-1 and forkhead box O1.
Figures
References
-
- Tisdale MJ. 2009. Mechanisms of cancer cachexia. Physiol Rev 89:381–410 - PubMed
-
- Evans WJ. 2010. Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr 91:1123S–1127S - PubMed
-
- Csibi A , Leibovitch MP , Cornille K , Tintignac LA , Leibovitch SA. 2009. MAFbx/Atrogin-1 controls the activity of the initiation factor eIF3-f in skeletal muscle atrophy by targeting multiple C-terminal lysines. J Biol Chem 284:4413–4421 - PubMed
-
- Csibi A , Tintignac LA , Leibovitch MP , Leibovitch SA. 2008. eIF3-f function in skeletal muscles: to stand at the crossroads of atrophy and hypertrophy. Cell Cycle 7:1698–1701 - PubMed
-
- Bodine SC , Latres E , Baumhueter S , Lai VK , Nunez L , Clarke BA , Poueymirou WT , Panaro FJ , Na E , Dharmarajan K , Pan ZQ , Valenzuela DM , DeChiara TM , Stitt TN , Yancopoulos GD , Glass DJ. 2001. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
