

Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy
- PMID: 21965549
- PMCID: PMC3248690
- DOI: 10.1161/CIRCGENETICS.111.961052
Homozygosity mapping and exome sequencing reveal GATAD1 mutation in autosomal recessive dilated cardiomyopathy
Abstract
Background: Dilated cardiomyopathy (DCM) is a heritable, genetically heterogeneous disorder that typically exhibits autosomal dominant inheritance. Genomic strategies enable discovery of novel, unsuspected molecular underpinnings of familial DCM. We performed genome-wide mapping and exome sequencing in a unique family wherein DCM segregated as an autosomal recessive (AR) trait.
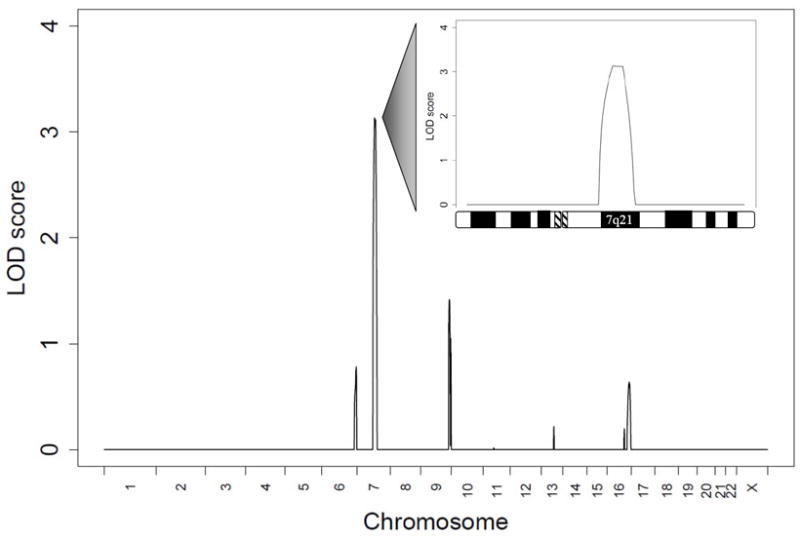
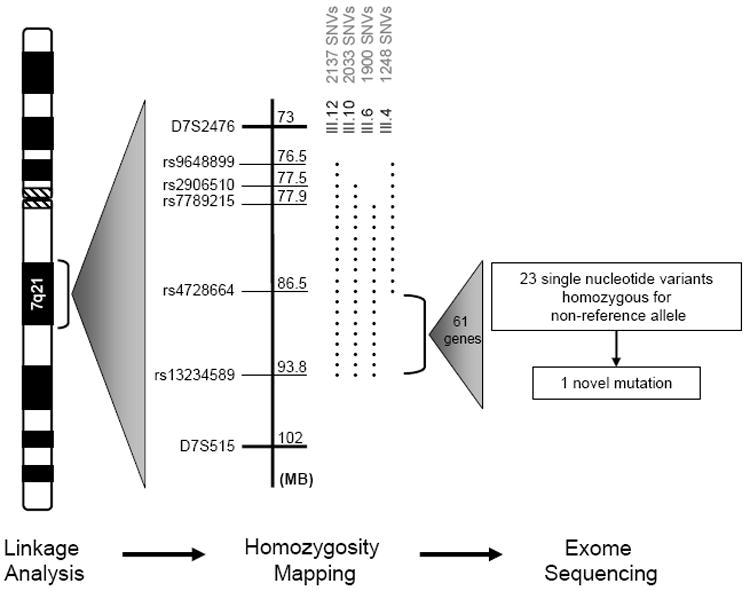
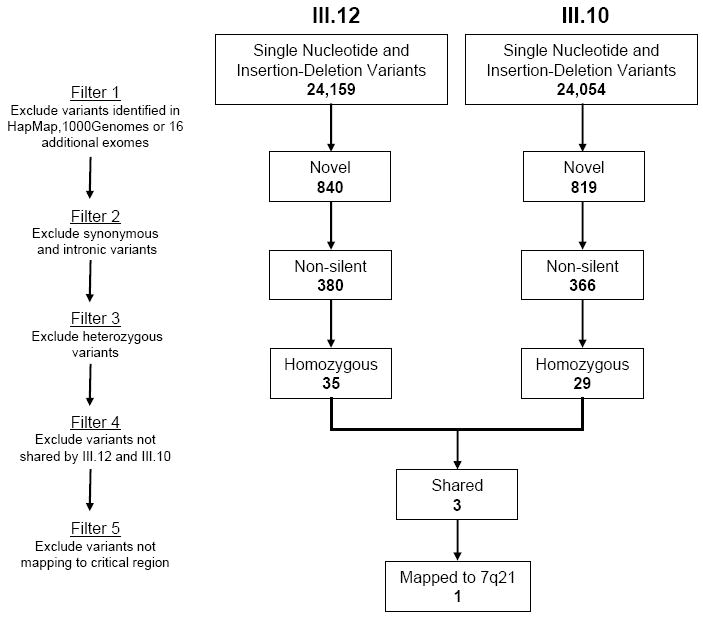
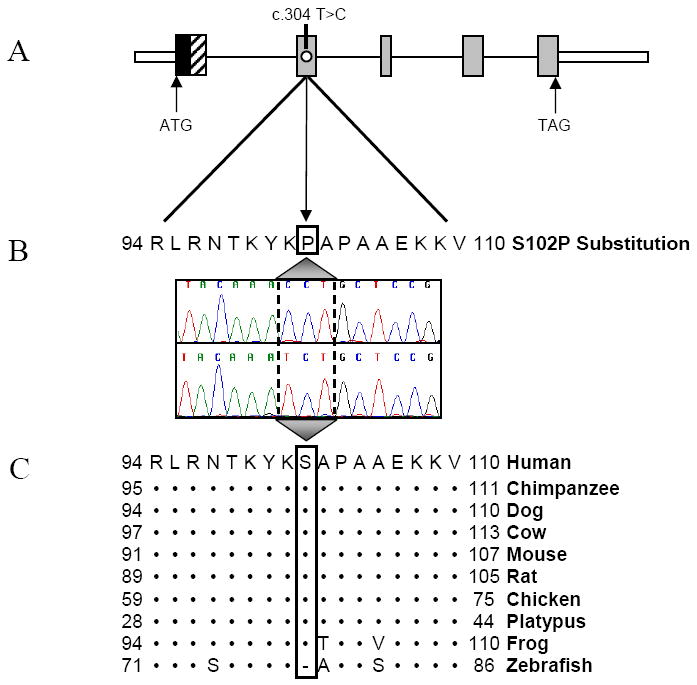
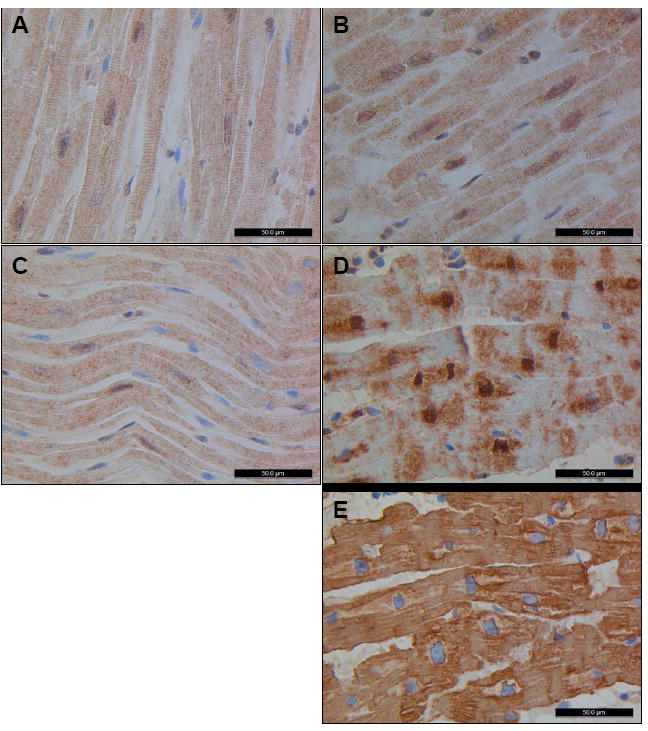
Methods and results: Echocardiography in 17 adult descendants of first cousins revealed DCM in 2 female siblings and idiopathic left ventricular enlargement in their brother. Genotyping and linkage analysis mapped an AR DCM locus to chromosome arm 7q21, which was validated and refined by high-density homozygosity mapping. Exome sequencing of the affected sisters was then used as a complementary strategy for mutation discovery. An iterative bioinformatics process was used to filter >40,000 genetic variants, revealing a single shared homozygous missense mutation localized to the 7q21 critical region. The mutation, absent in HapMap, 1000 Genomes, and 474 ethnically matched controls, altered a conserved residue of GATAD1, encoding GATA zinc finger domain-containing protein 1. Thirteen relatives were heterozygous mutation carriers with no evidence of myocardial disease, even at advanced ages. Immunohistochemistry demonstrated nuclear localization of GATAD1 in left ventricular myocytes, yet subcellular expression and nuclear morphology were aberrant in the proband.
Conclusions: Linkage analysis and exome sequencing were used as synergistic genomic strategies to identify GATAD1 as a gene for AR DCM. GATAD1 binds to a histone modification site that regulates gene expression. Consistent with murine DCM caused by genetic disruption of histone deacetylases, the data implicate an inherited basis for epigenetic dysregulation in human heart failure.
Conflict of interest statement
Figures

Similar articles
-
A substitution mutation in cardiac ubiquitin ligase, FBXO32, is associated with an autosomal recessive form of dilated cardiomyopathy.BMC Med Genet. 2016 Jan 14;17:3. doi: 10.1186/s12881-016-0267-5. BMC Med Genet. 2016. PMID: 26768247 Free PMC article.
-
Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy.J Am Coll Cardiol. 2009 Sep 1;54(10):930-41. doi: 10.1016/j.jacc.2009.05.038. J Am Coll Cardiol. 2009. PMID: 19712804 Free PMC article.
-
Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy.Circ Cardiovasc Genet. 2013 Apr;6(2):144-53. doi: 10.1161/CIRCGENETICS.111.000062. Epub 2013 Feb 15. Circ Cardiovasc Genet. 2013. PMID: 23418287 Free PMC article.
-
TNNI3K mutation in familial syndrome of conduction system disease, atrial tachyarrhythmia and dilated cardiomyopathy.Hum Mol Genet. 2014 Nov 1;23(21):5793-804. doi: 10.1093/hmg/ddu297. Epub 2014 Jun 11. Hum Mol Genet. 2014. PMID: 24925317 Free PMC article.
-
Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy.Circ Cardiovasc Genet. 2013 Aug;6(4):317-26. doi: 10.1161/CIRCGENETICS.113.000011. Epub 2013 Jul 16. Circ Cardiovasc Genet. 2013. PMID: 23861363 Free PMC article.
Cited by
-
Animal Models to Study Cardiac Arrhythmias.Circ Res. 2022 Jun 10;130(12):1926-1964. doi: 10.1161/CIRCRESAHA.122.320258. Epub 2022 Jun 9. Circ Res. 2022. PMID: 35679367 Free PMC article. Review.
-
Exome sequencing greatly expedites the progressive research of Mendelian diseases.Front Med. 2014 Mar;8(1):42-57. doi: 10.1007/s11684-014-0303-9. Epub 2014 Jan 3. Front Med. 2014. PMID: 24384736 Review.
-
Epigenetic regulation of puberty via Zinc finger protein-mediated transcriptional repression.Nat Commun. 2015 Dec 16;6:10195. doi: 10.1038/ncomms10195. Nat Commun. 2015. PMID: 26671628 Free PMC article.
-
Modeling GATAD1-Associated Dilated Cardiomyopathy in Adult Zebrafish.J Cardiovasc Dev Dis. 2016 Mar;3(1):6. doi: 10.3390/jcdd3010006. Epub 2016 Jan 26. J Cardiovasc Dev Dis. 2016. PMID: 28955713 Free PMC article.
-
Loss of GATAD1 in cardiomyocyte does not cause cardiomyopathy in mice.J Mol Histol. 2024 Dec 6;56(1):33. doi: 10.1007/s10735-024-10297-z. J Mol Histol. 2024. PMID: 39641830 Free PMC article.
References
-
- Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. - PubMed
-
- Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. - PubMed
-
- Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
