

APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells
- PMID: 21966267
- PMCID: PMC3178557
- DOI: 10.1371/journal.ppat.1002221
APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells
Abstract
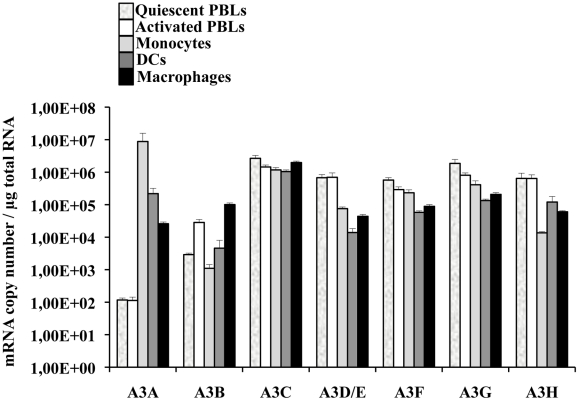
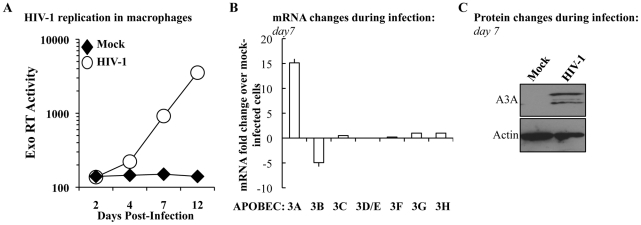
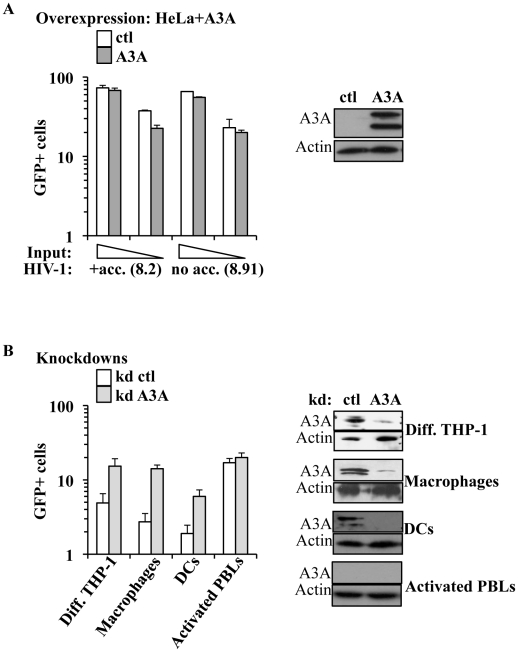
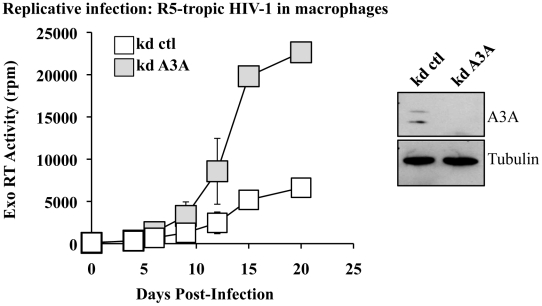
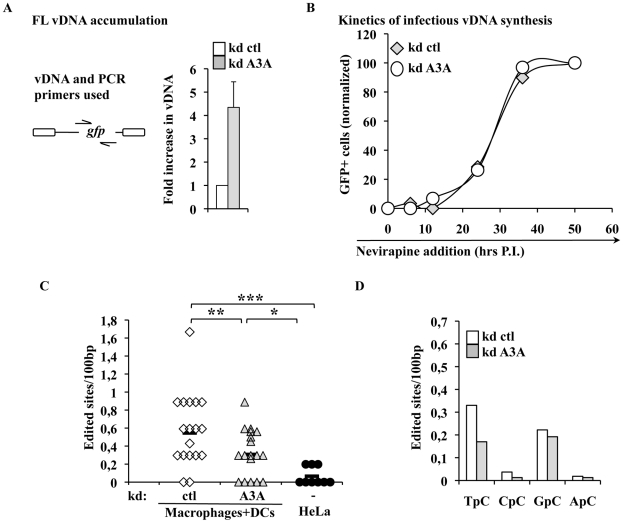
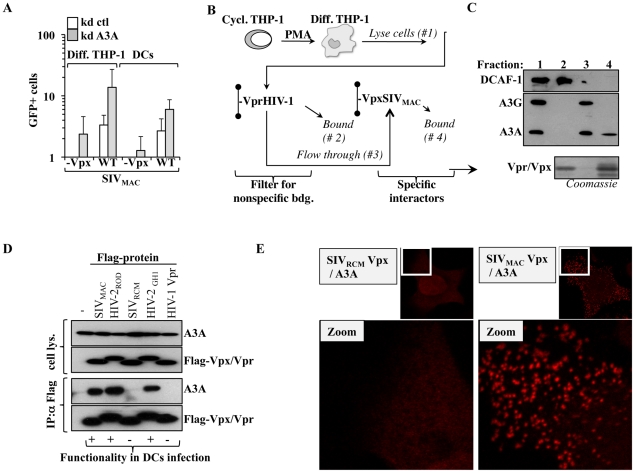
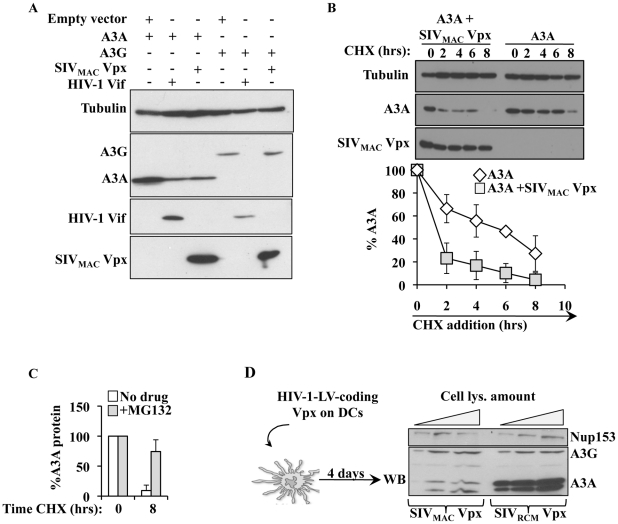
Myeloid cells play numerous roles in HIV-1 pathogenesis serving as a vehicle for viral spread and as a viral reservoir. Yet, cells of this lineage generally resist HIV-1 infection when compared to cells of other lineages, a phenomenon particularly acute during the early phases of infection. Here, we explore the role of APOBEC3A on these steps. APOBEC3A is a member of the APOBEC3 family that is highly expressed in myeloid cells, but so far lacks a known antiviral effect against retroviruses. Using ectopic expression of APOBEC3A in established cell lines and specific silencing in primary macrophages and dendritic cells, we demonstrate that the pool of APOBEC3A in target cells inhibits the early phases of HIV-1 infection and the spread of replication-competent R5-tropic HIV-1, specifically in cells of myeloid origins. In these cells, APOBEC3A affects the amount of vDNA synthesized over the course of infection. The susceptibility to the antiviral effect of APOBEC3A is conserved among primate lentiviruses, although the viral protein Vpx coded by members of the SIV(SM)/HIV-2 lineage provides partial protection from APOBEC3A during infection. Our results indicate that APOBEC3A is a previously unrecognized antiviral factor that targets primate lentiviruses specifically in myeloid cells and that acts during the early phases of infection directly in target cells. The findings presented here open up new venues on the role of APOBEC3A during HIV infection and pathogenesis, on the role of the cellular context in the regulation of the antiviral activities of members of the APOBEC3 family and more generally on the natural functions of APOBEC3A.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. - PubMed
-
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, et al. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. - PubMed
-
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
