

Development of novel CH223191-based antagonists of the aryl hydrocarbon receptor
- PMID: 21967751
- PMCID: PMC3250110
- DOI: 10.1124/mol.111.073643
Development of novel CH223191-based antagonists of the aryl hydrocarbon receptor
Abstract
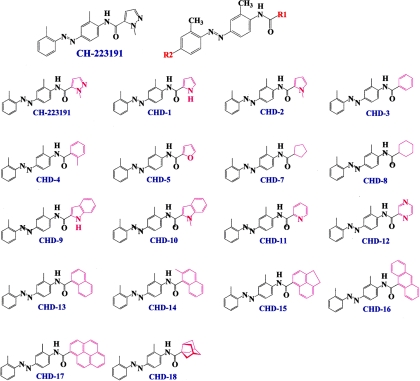
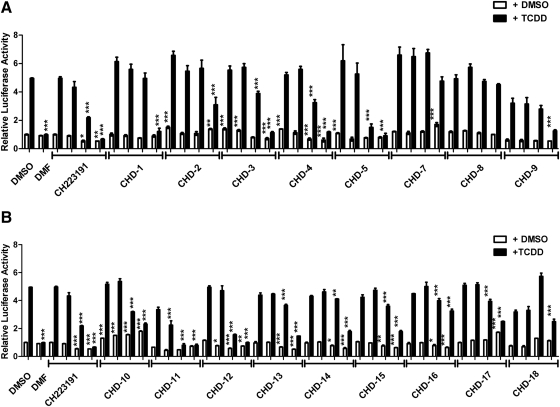
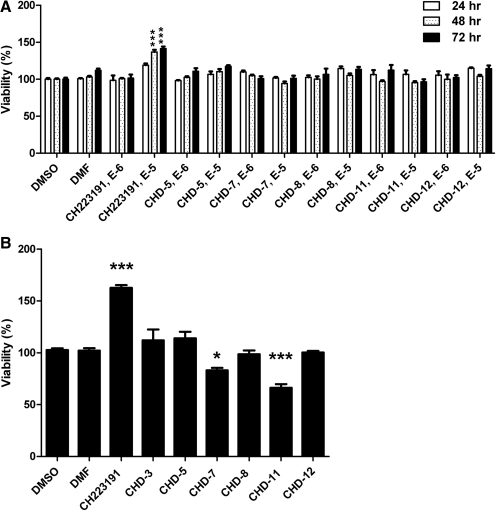
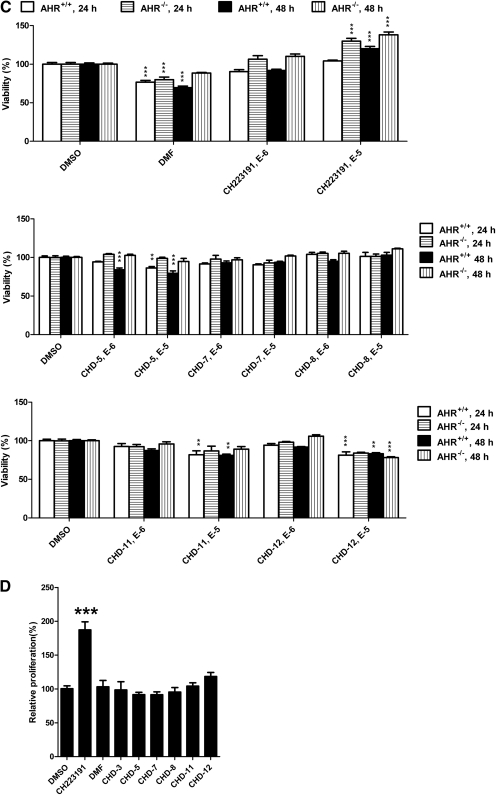
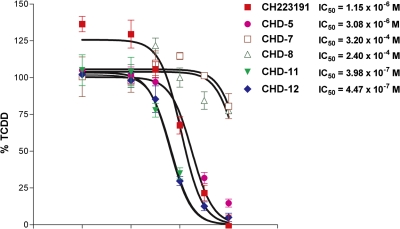
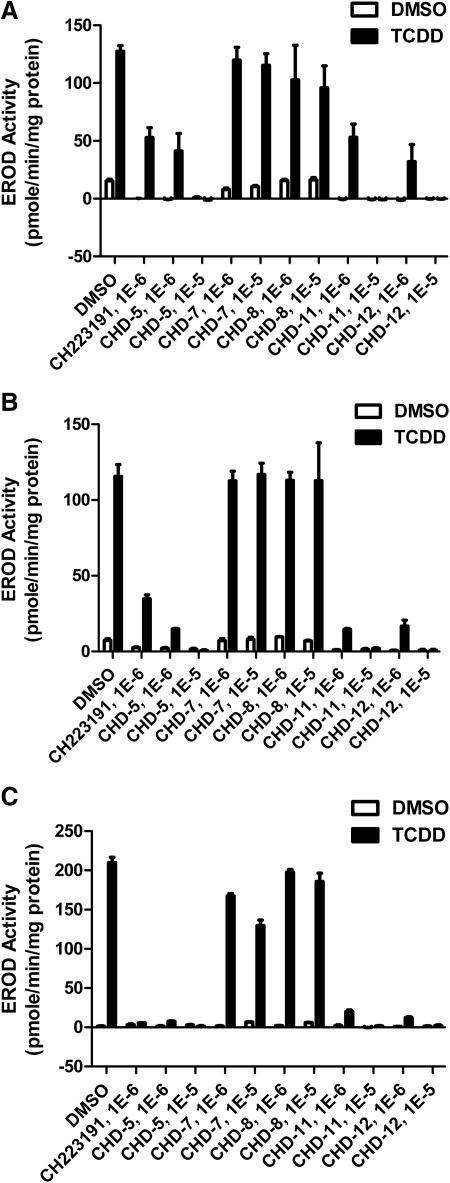
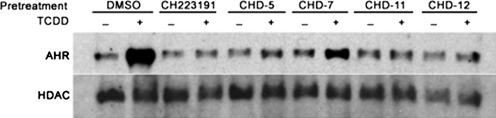
Aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that regulates genes involved in drug/xenobiotic metabolism, cell cycle progression, cell fate determination, immune function, and inflammatory response. Increasing evidence that AHR plays a role in the pathophysiology of a number of human disease states is driving the need for improved pharmacological tools to be used for understanding the in vivo impact of AHR modulation. In this study, we have characterized and used structure-activity relationship analyses of a newly synthesized library of derivatives of the potent AHR antagonist 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH223191). Initial screening of these compounds revealed that those bearing groups with strong electronegativity at the R1 position (i.e., CHD-5, CHD-11, and CHD-12) versus those that are more electron-poor at this position (i.e., CHD-7 and CHD-8) elicited the most potent AHR antagonistic properties. The ability of these derivatives to inhibit agonist (2,3,7,8-tetrachlorodibenzo-p-dioxin) binding, nuclear translocation of AHR, and agonist-induced enzyme activity also were determined and support the initial findings. Furthermore, CH223191, but not CHD-5, CHD-11, or CHD-12, was found to exhibit AHR-independent proproliferative properties. These results contribute to our understanding of the structural requirements of potent AHR antagonists and the development of effective pharmacological tools to be used for studying the pathophysiological role of AHR.
Figures
References
-
- Denison MS, Nagy SR. (2003) Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 43:309–334 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
