

Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages
- PMID: 21969549
- PMCID: PMC3193184
- DOI: 10.1073/pnas.1111245108
Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages
Abstract
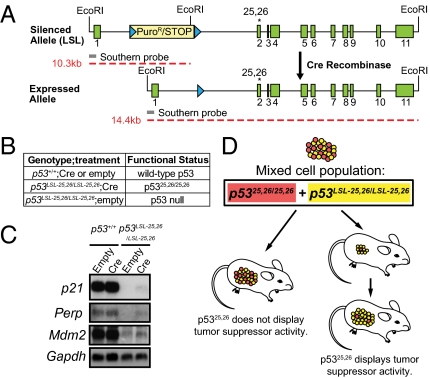
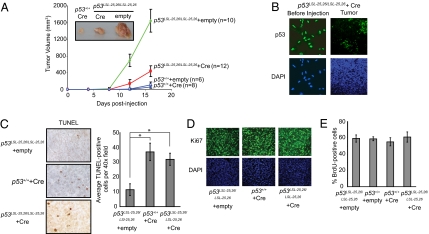
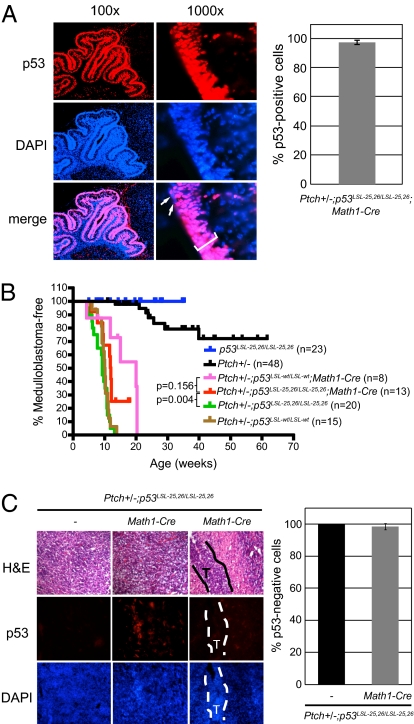
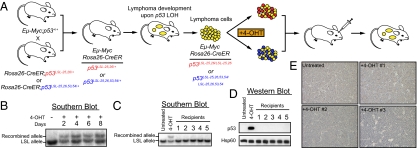
Over half of all human cancers, of a wide variety of types, sustain mutations in the p53 tumor suppressor gene. Although p53 limits tumorigenesis through the induction of apoptosis or cell cycle arrest, its molecular mechanism of action in tumor suppression has been elusive. The best-characterized p53 activity in vitro is as a transcriptional activator, but the identification of numerous additional p53 biochemical activities in vitro has made it unclear which mechanism accounts for tumor suppression. Here, we assess the importance of transcriptional activation for p53 tumor suppression function in vivo in several tissues, using a knock-in mouse strain expressing a p53 mutant compromised for transcriptional activation, p53(25,26). p53(25,26) is severely impaired for the transactivation of numerous classical p53 target genes, including p21, Noxa, and Puma, but it retains the ability to activate a small subset of p53 target genes, including Bax. Surprisingly, p53(25,26) can nonetheless suppress tumor growth in cancers derived from the epithelial, mesenchymal, central nervous system, and lymphoid lineages. Therefore, full transactivation of most p53 target genes is dispensable for p53 tumor suppressor function in a range of tissue types. In contrast, a transcriptional activation mutant that is completely defective for transactivation, p53(25,26,53,54), fails to suppress tumor development. These findings demonstrate that transcriptional activation is indeed broadly critical for p53 tumor suppressor function, although this requirement reflects the limited transcriptional activity observed with p53(25,26) rather than robust transactivation of a full complement of p53 target genes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell. 2009;137:413–431. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
