

Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes
- PMID: 21971515
- PMCID: PMC3217123
- DOI: 10.1016/j.bbabio.2011.09.011
Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes
Abstract
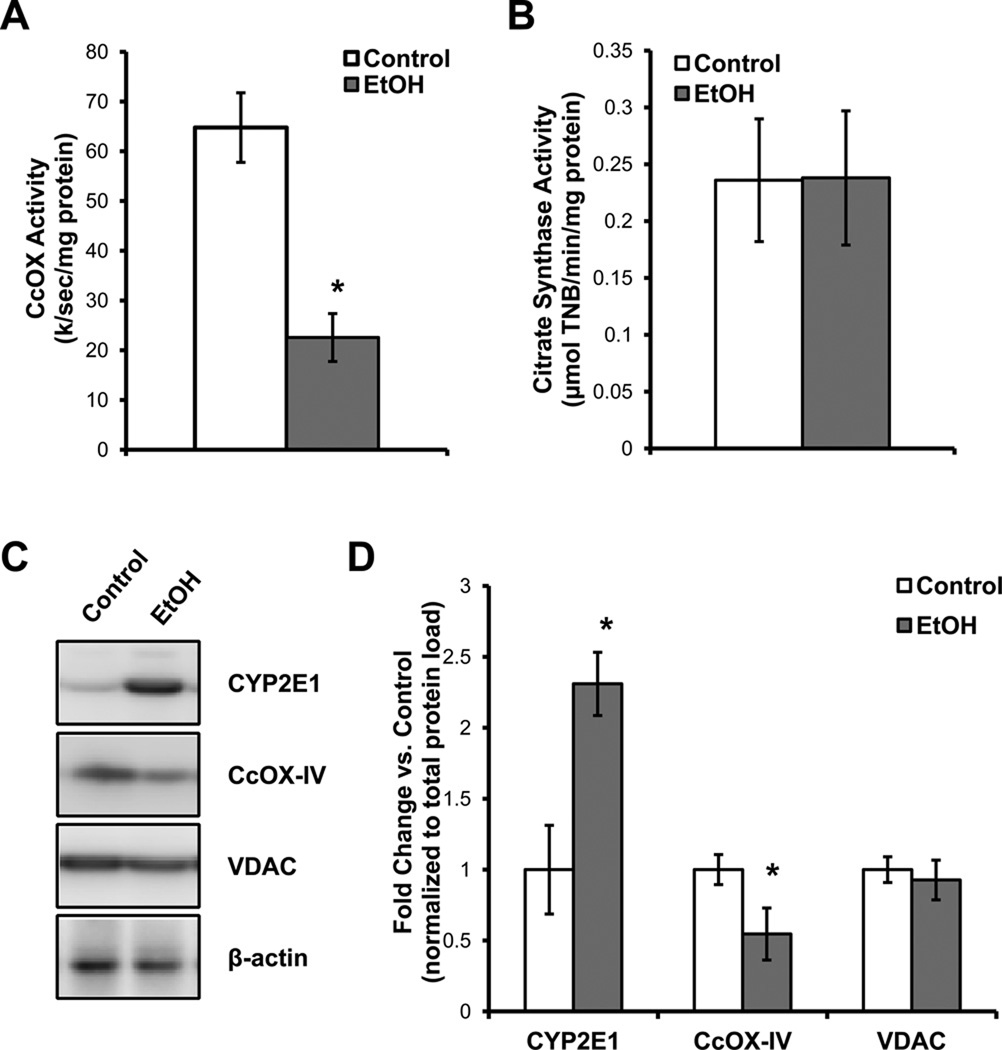
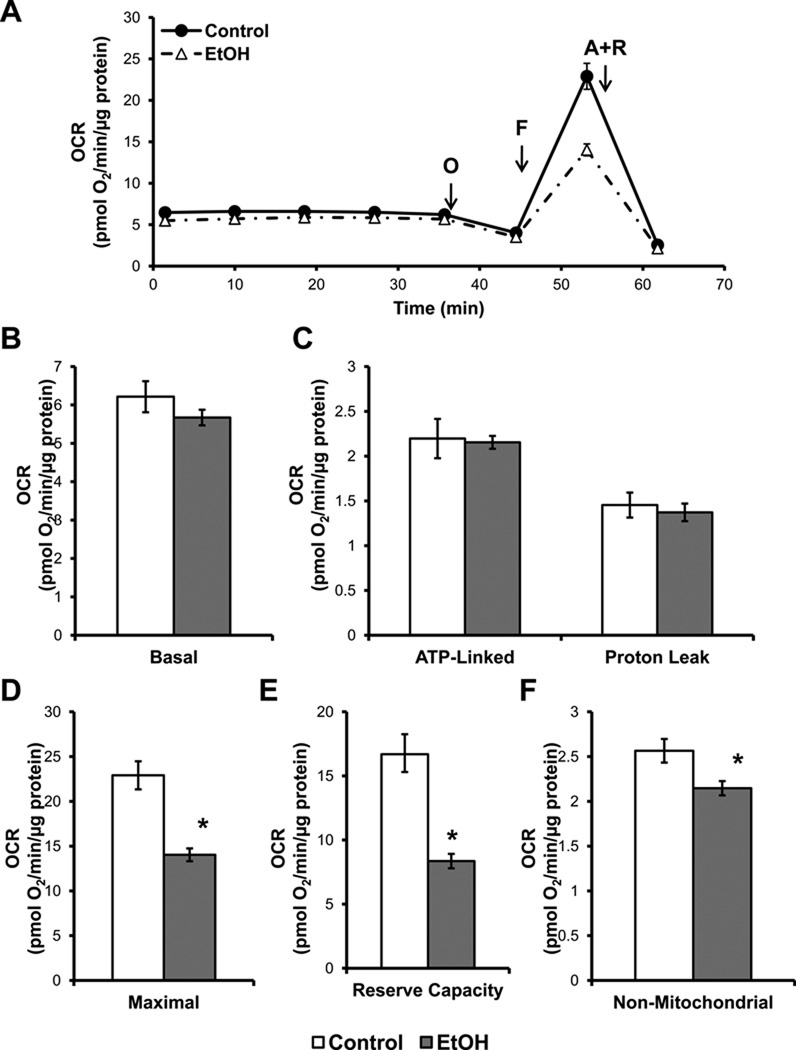
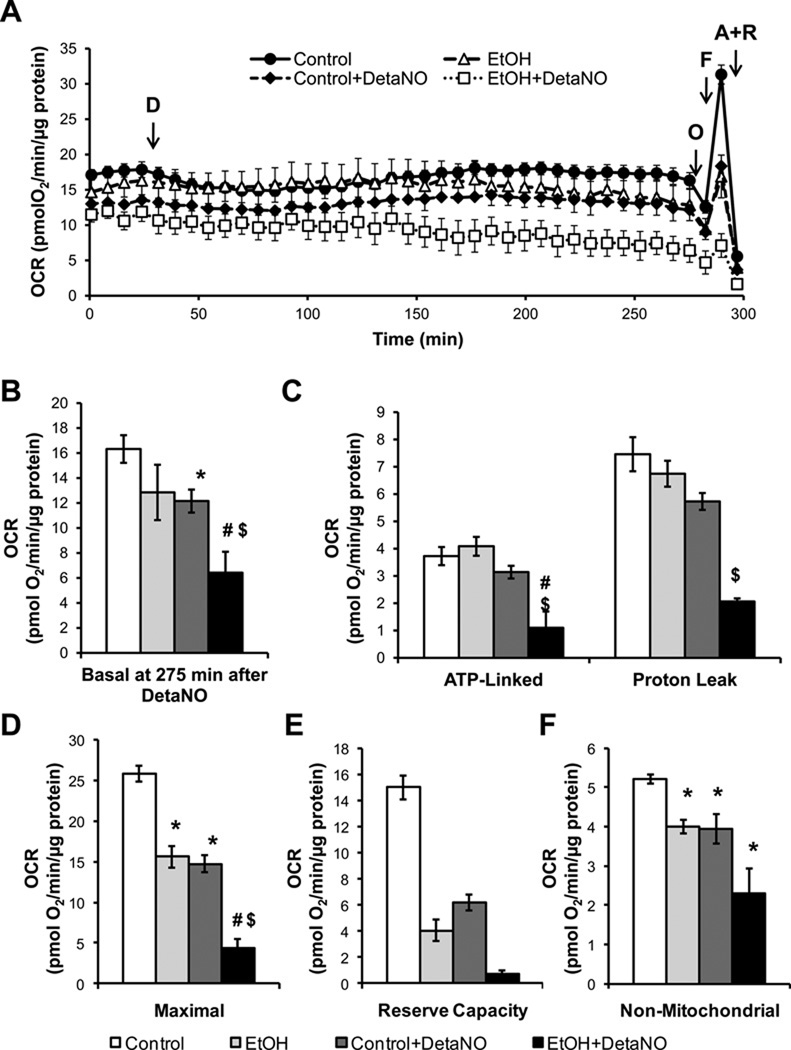
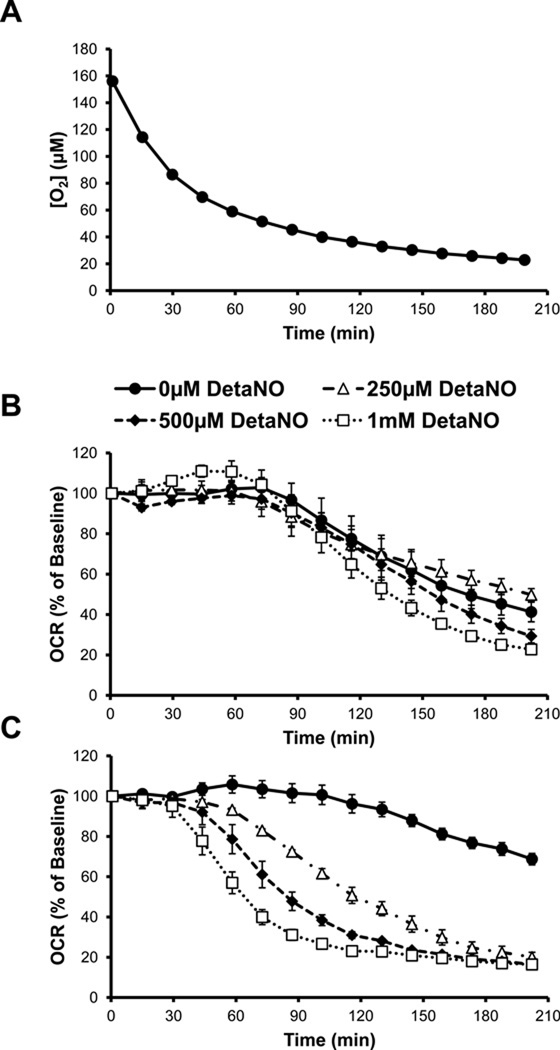
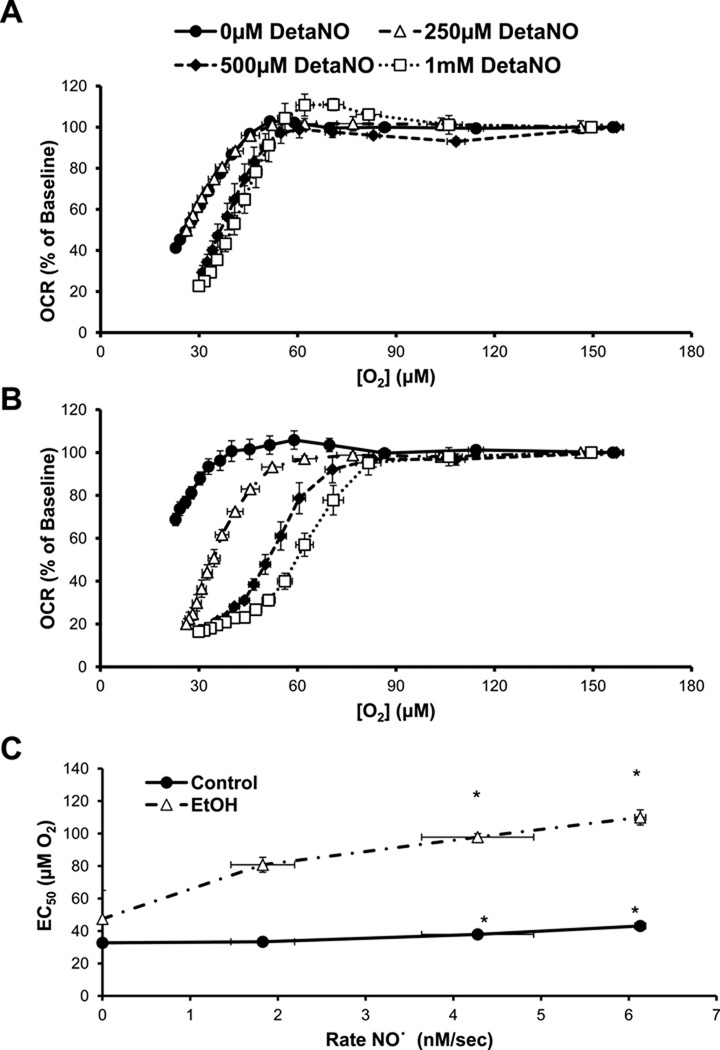
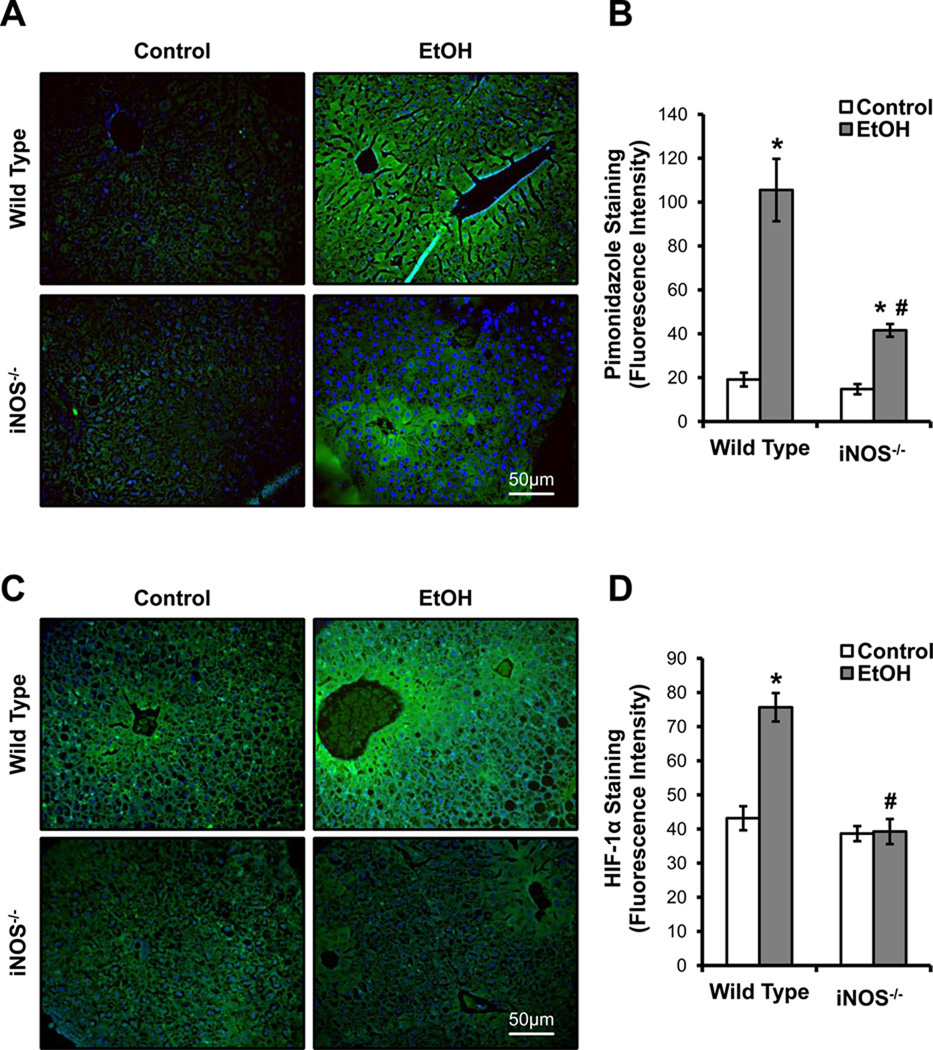
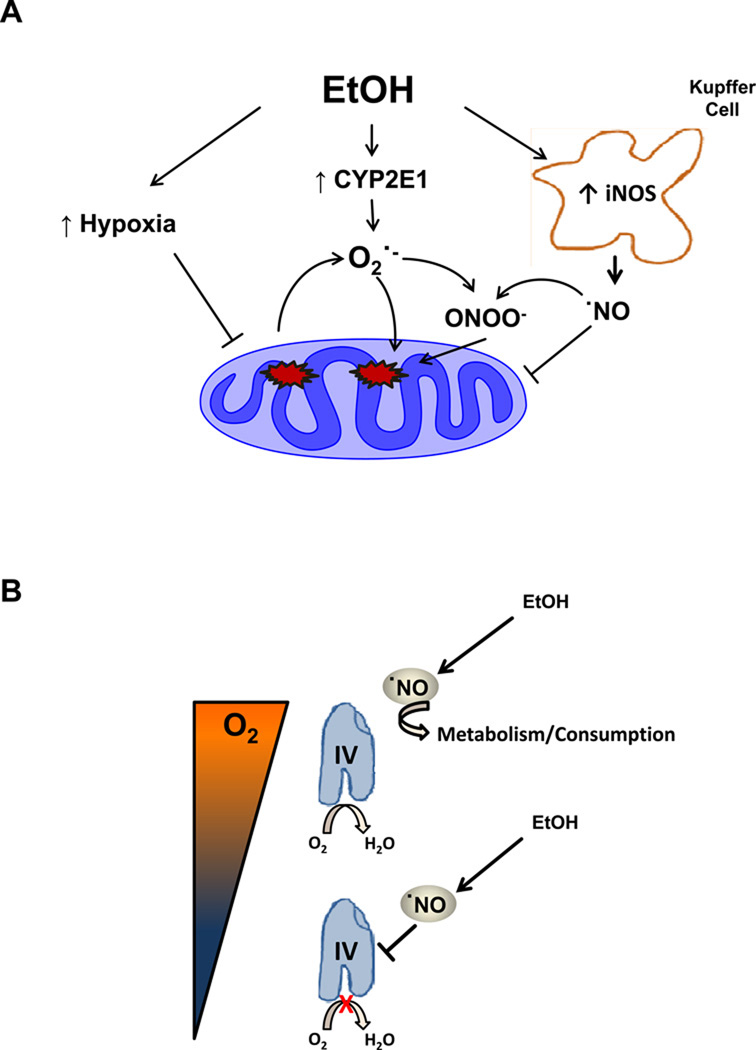
Chronic alcohol consumption results in hepatotoxicity, steatosis, hypoxia, increased expression of inducible nitric oxide synthase (iNOS) and decreased activities of mitochondrial respiratory enzymes. The impact of these changes on cellular respiration and their interaction in a cellular setting is not well understood. In the present study we tested the hypothesis that nitric oxide (NO)-dependent modulation of cellular respiration and the sensitivity to hypoxic stress is increased following chronic alcohol consumption. This is important since NO has been shown to regulate mitochondrial function through its interaction with cytochrome c oxidase, although at higher concentrations, and in combination with reactive oxygen species, can result in mitochondrial dysfunction. We found that hepatocytes isolated from alcohol-fed rats had decreased mitochondrial bioenergetic reserve capacity and were more sensitive to NO-dependent inhibition of respiration under room air and hypoxic conditions. We reasoned that this would result in greater hypoxic stress in vivo, and to test this, wild-type and iNOS(-/-) mice were administered alcohol-containing diets. Chronic alcohol consumption resulted in liver hypoxia in the wild-type mice and increased levels of hypoxia-inducible factor 1 α in the peri-venular region of the liver lobule. These effects were attenuated in the alcohol-fed iNOS(-/-) mice suggesting that increased mitochondrial sensitivity to NO and reactive nitrogen species in hepatocytes and iNOS plays a critical role in determining the response to hypoxic stress in vivo. These data support the concept that the combined effects of NO and ethanol contribute to an increased susceptibility to hypoxia and the deleterious effects of alcohol consumption on liver.
2011 Elsevier B.V. All rights reserved.
Figures
References
-
- Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling: from physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–2531. - PubMed
-
- Xu W, Charles IG, Moncada S. Nitric oxide: orchestrating hypoxia regulation through mitochondrial respiration and the endoplasmic reticulum stress response. Cell Res. 2005;15:63–65. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
