

Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene
- PMID: 21972111
- PMCID: PMC3724403
- DOI: 10.1002/humu.21426
Nonsense mutation-associated Becker muscular dystrophy: interplay between exon definition and splicing regulatory elements within the DMD gene
Abstract
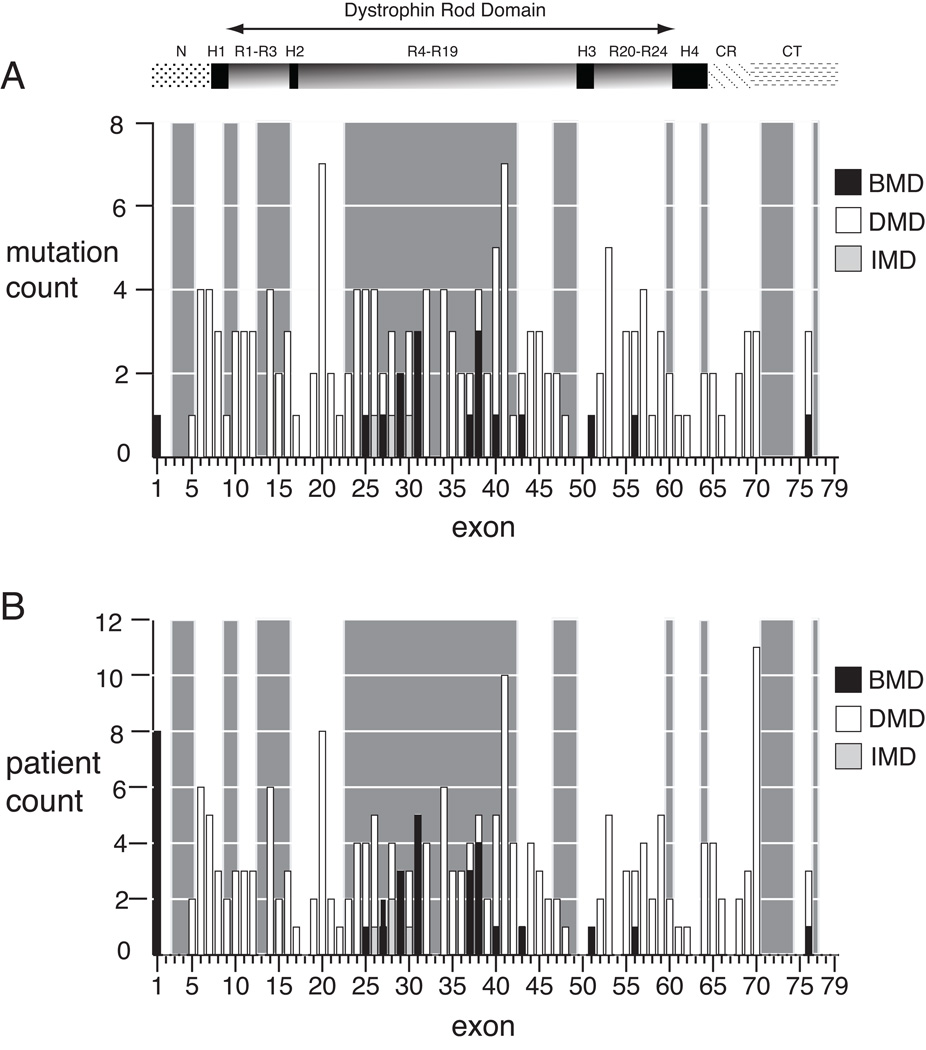
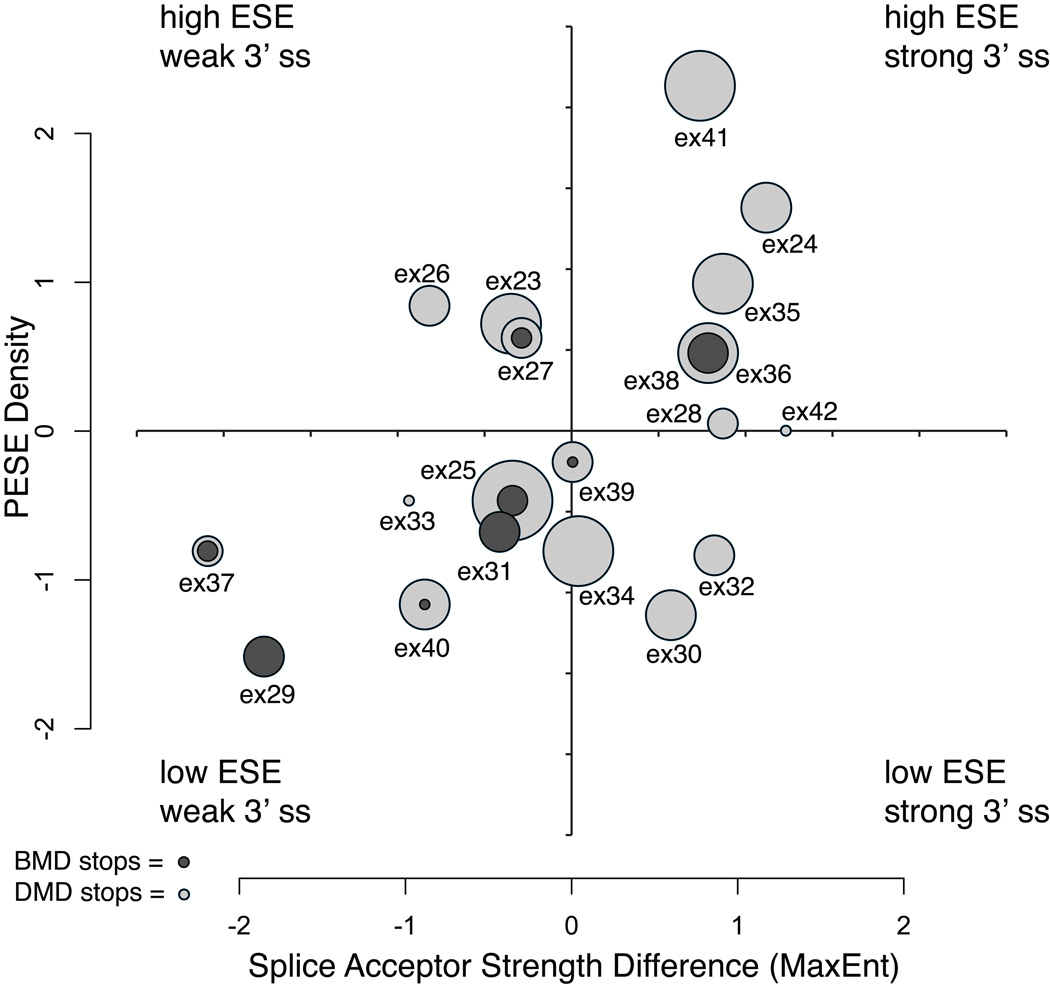
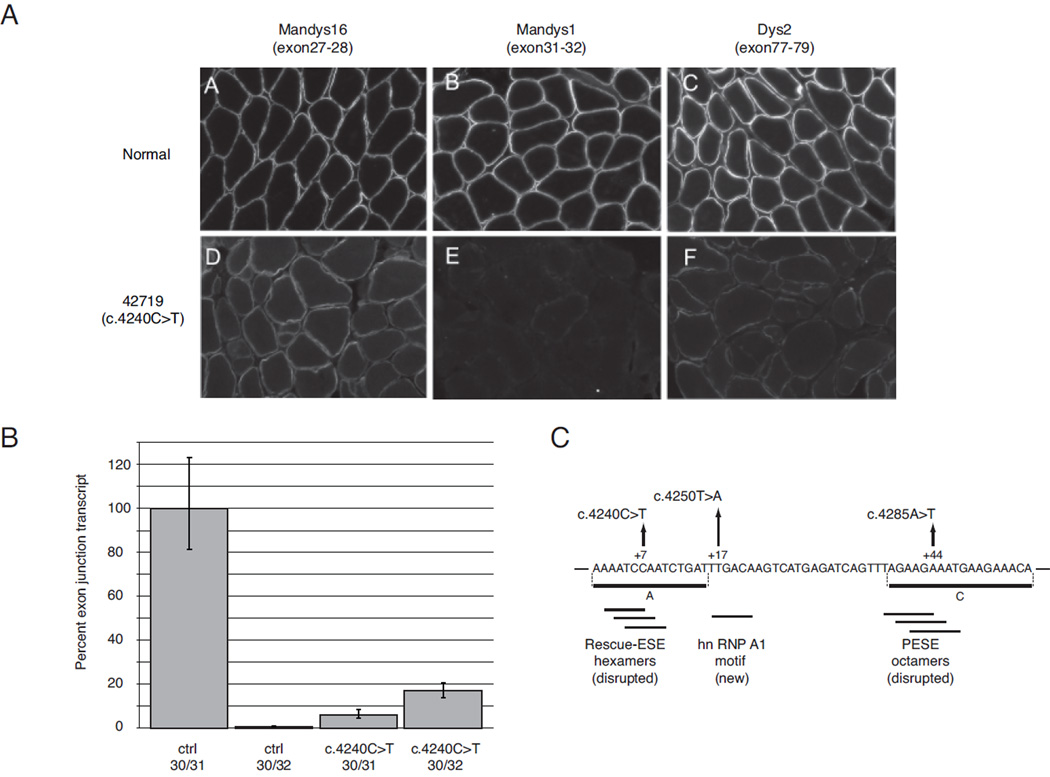
Nonsense mutations are usually predicted to function as null alleles due to premature termination of protein translation. However, nonsense mutations in the DMD gene, encoding the dystrophin protein, have been associated with both the severe Duchenne Muscular Dystrophy (DMD) and milder Becker Muscular Dystrophy (BMD) phenotypes. In a large survey, we identified 243 unique nonsense mutations in the DMD gene, and for 210 of these we could establish definitive phenotypes. We analyzed the reading frame predicted by exons flanking those in which nonsense mutations were found, and present evidence that nonsense mutations resulting in BMD likely do so by inducing exon skipping, confirming that exonic point mutations affecting exon definition have played a significant role in determining phenotype. We present a new model based on the combination of exon definition and intronic splicing regulatory elements for the selective association of BMD nonsense mutations with a subset of DMD exons prone to mutation-induced exon skipping.
© 2011 Wiley-Liss, Inc.
Figures

References
-
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 2010;465(7294):53–59. - PubMed
-
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30(4):377–384. - PubMed
-
- Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, Hart KJ, Tuohy T, White S, den Dunnen JT and others. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134(3):295–298. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
