

Metabolic stress, reactive oxygen species, and arrhythmia
- PMID: 21978629
- PMCID: PMC3264827
- DOI: 10.1016/j.yjmcc.2011.09.018
Metabolic stress, reactive oxygen species, and arrhythmia
Abstract
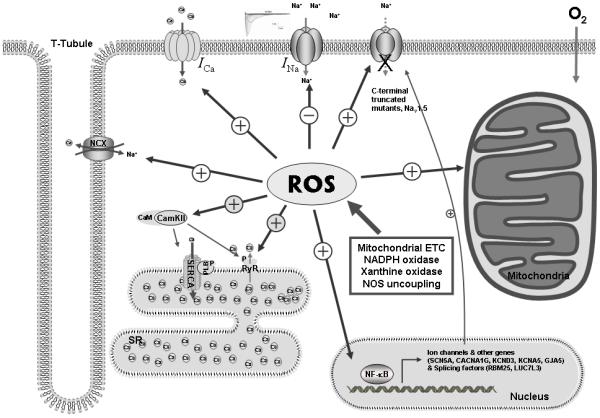
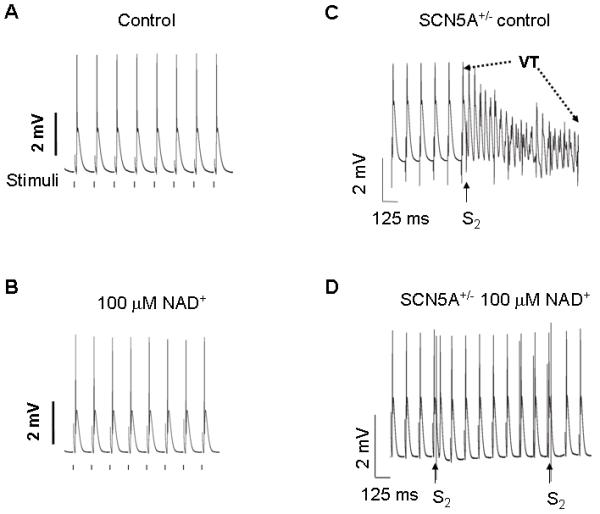
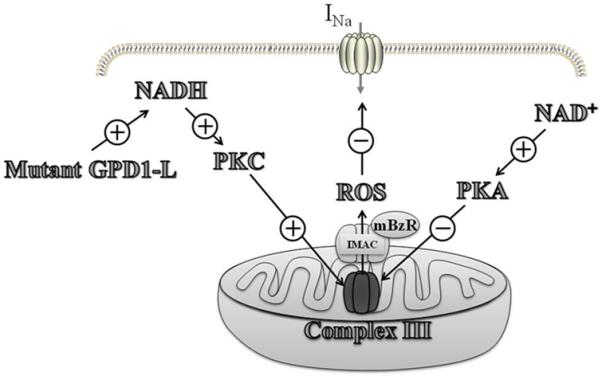
Cardiac arrhythmias can cause sudden cardiac death (SCD) and add to the current heart failure (HF) health crisis. Nevertheless, the pathological processes underlying arrhythmias are unclear. Arrhythmic conditions are associated with systemic and cardiac oxidative stress caused by reactive oxygen species (ROS). In excitable cardiac cells, ROS regulate both cellular metabolism and ion homeostasis. Increasing evidence suggests that elevated cellular ROS can cause alterations of the cardiac sodium channel (Na(v)1.5), abnormal Ca(2+) handling, changes of mitochondrial function, and gap junction remodeling, leading to arrhythmogenesis. This review summarizes our knowledge of the mechanisms by which ROS may cause arrhythmias and discusses potential therapeutic strategies to prevent arrhythmias by targeting ROS and its consequences. This article is part of a Special Issue entitled "Local Signaling in Myocytes".
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- Stecker EC, Vickers C, Waltz J, Socoteanu C, John BT, Mariani R, et al. Population-based analysis of sudden cardiac death with and without left ventricular systolic dysfunction: two-year findings from the Oregon Sudden Unexpected Death Study. J Am Coll Cardiol. 2006;47:1161–6. - PubMed
-
- Arking DE, Chugh SS, Chakravarti A, Spooner PM. Genomics in sudden cardiac death. Circ Res. 2004;94:712–23. - PubMed
-
- Fuster V, Ryden LE, Cannom DS, Crijns HJ, Curtis AB, Ellenbogen KA, et al. ACC/AHA/ESC 2006 Guidelines for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association task force on practice guidelines and the European Society of Cardiology Committee for practice guidelines (Writing committee to revise the 2001 Guidelines for the management of patients with atrial fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Circulation. 2006;114:e257–e354. - PubMed
-
- Kannel WB, Abbott RD, Savage DD, McNamara PM. Epidemiologic features of chronic atrial fibrillation: the Framingham study. N Engl J Med. 1982;306:1018–22. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
