

Understanding the structural ensembles of a highly extended disordered protein
- PMID: 21979461
- PMCID: PMC3645981
- DOI: 10.1039/c1mb05243h
Understanding the structural ensembles of a highly extended disordered protein
Abstract
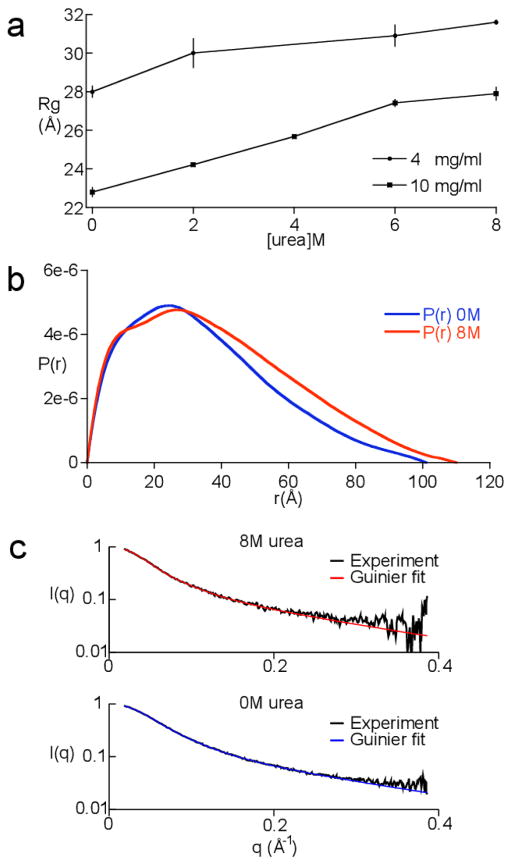
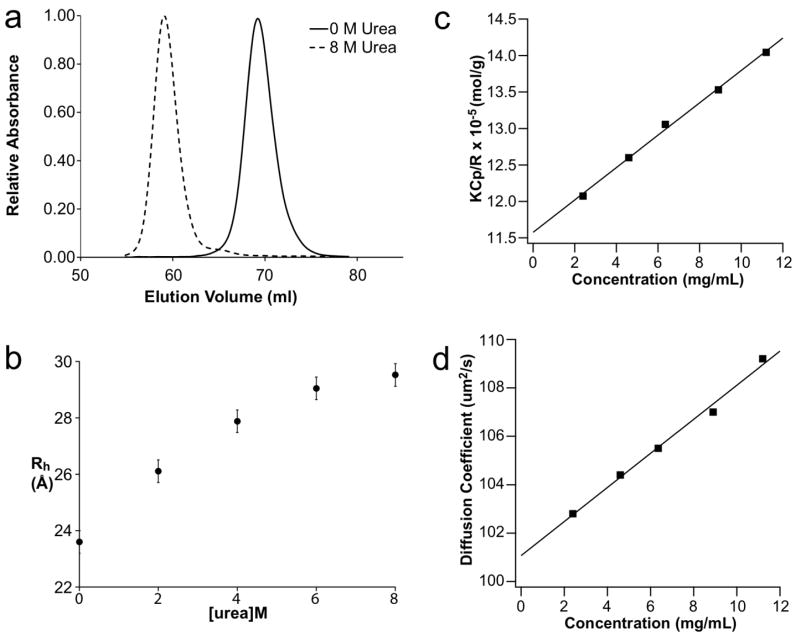
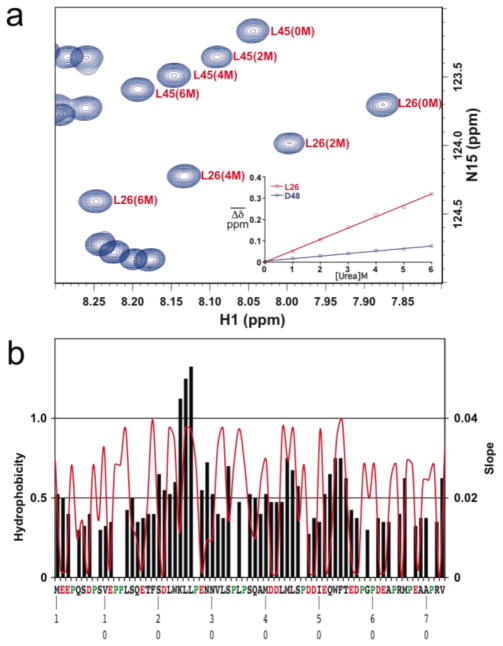
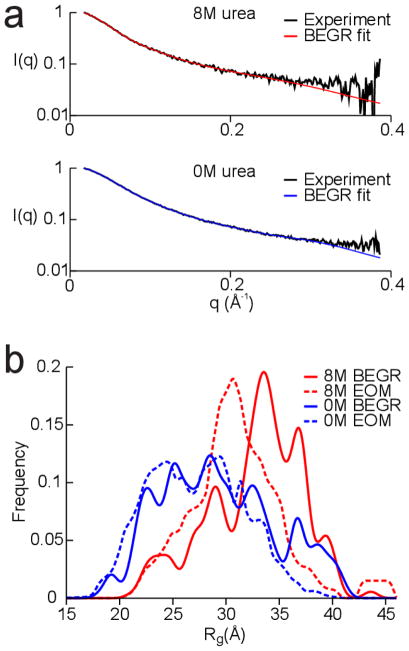
Developing a comprehensive description of the equilibrium structural ensembles for intrinsically disordered proteins (IDPs) is essential to understanding their function. The p53 transactivation domain (p53TAD) is an IDP that interacts with multiple protein partners and contains numerous phosphorylation sites. Multiple techniques were used to investigate the equilibrium structural ensemble of p53TAD in its native and chemically unfolded states. The results from these experiments show that the native state of p53TAD has dimensions similar to a classical random coil while the chemically unfolded state is more extended. To investigate the molecular properties responsible for this behavior, a novel algorithm that generates diverse and unbiased structural ensembles of IDPs was developed. This algorithm was used to generate a large pool of plausible p53TAD structures that were reweighted to identify a subset of structures with the best fit to small angle X-ray scattering data. High weight structures in the native state ensemble show features that are localized to protein binding sites and regions with high proline content. The features localized to the protein binding sites are mostly eliminated in the chemically unfolded ensemble; while, the regions with high proline content remain relatively unaffected. Data from NMR experiments support these results, showing that residues from the protein binding sites experience larger environmental changes upon unfolding by urea than regions with high proline content. This behavior is consistent with the urea-induced exposure of nonpolar and aromatic side-chains in the protein binding sites that are partially excluded from solvent in the native state ensemble.
Figures
References
-
- Wright PE, Dyson HJ. J Mol Biol. 1999;293:321–331. - PubMed
-
- Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker AK. In: Protein Folding Handbook. Buchner J, Kiefhaber T, editors. Vol. 3. WILEY-VCH; Darmstadt: 2005. pp. 275–357.
-
- Vendruscolo M. Curr Opin Struct Biol. 2007;17:15–20. - PubMed
-
- Uversky VN. Eur J Biochem. 2002;269:2–12. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous