

Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels
- PMID: 21980127
- PMCID: PMC3234531
- DOI: 10.1161/CIRCRESAHA.111.240242
Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels
Abstract
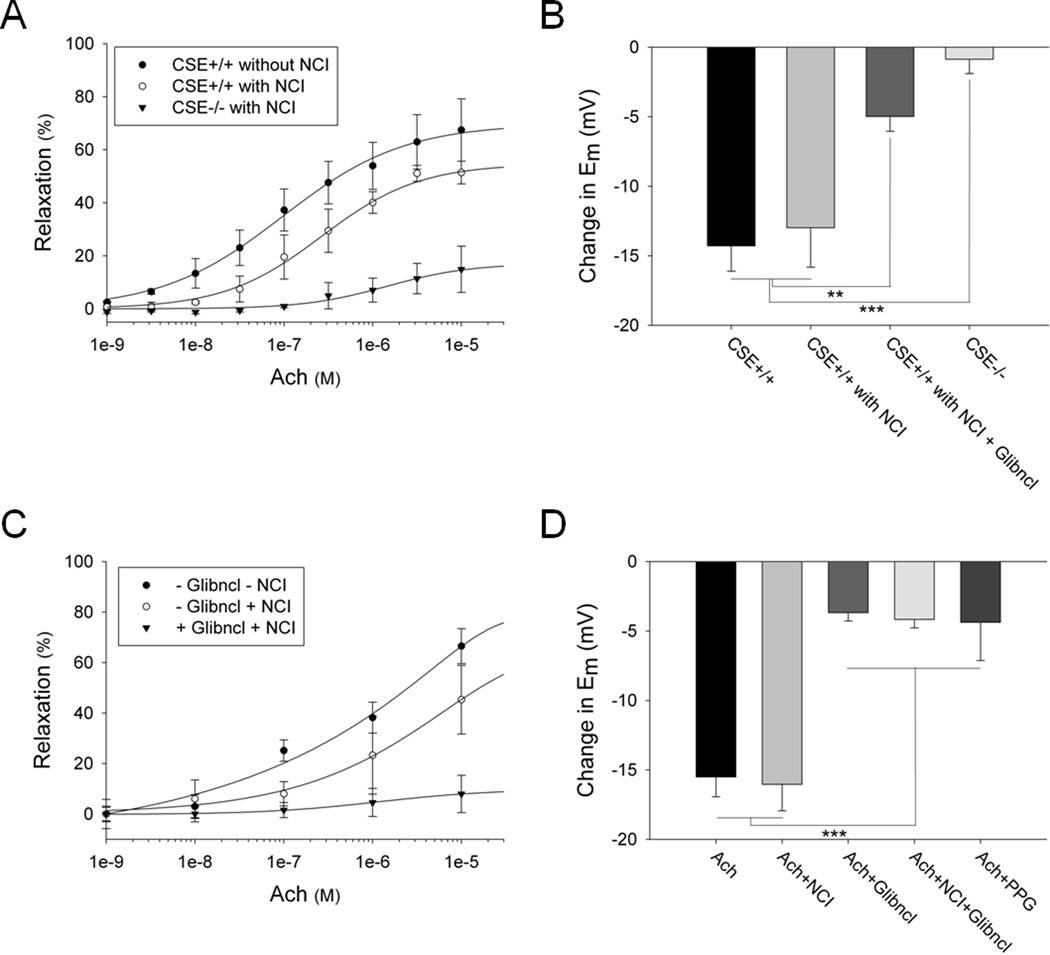
Rationale: Nitric oxide, the classic endothelium-derived relaxing factor (EDRF), acts through cyclic GMP and calcium without notably affecting membrane potential. A major component of EDRF activity derives from hyperpolarization and is termed endothelium-derived hyperpolarizing factor (EDHF). Hydrogen sulfide (H(2)S) is a prominent EDRF, since mice lacking its biosynthetic enzyme, cystathionine γ-lyase (CSE), display pronounced hypertension with deficient vasorelaxant responses to acetylcholine.
Objective: The purpose of this study was to determine if H(2)S is a major physiological EDHF.
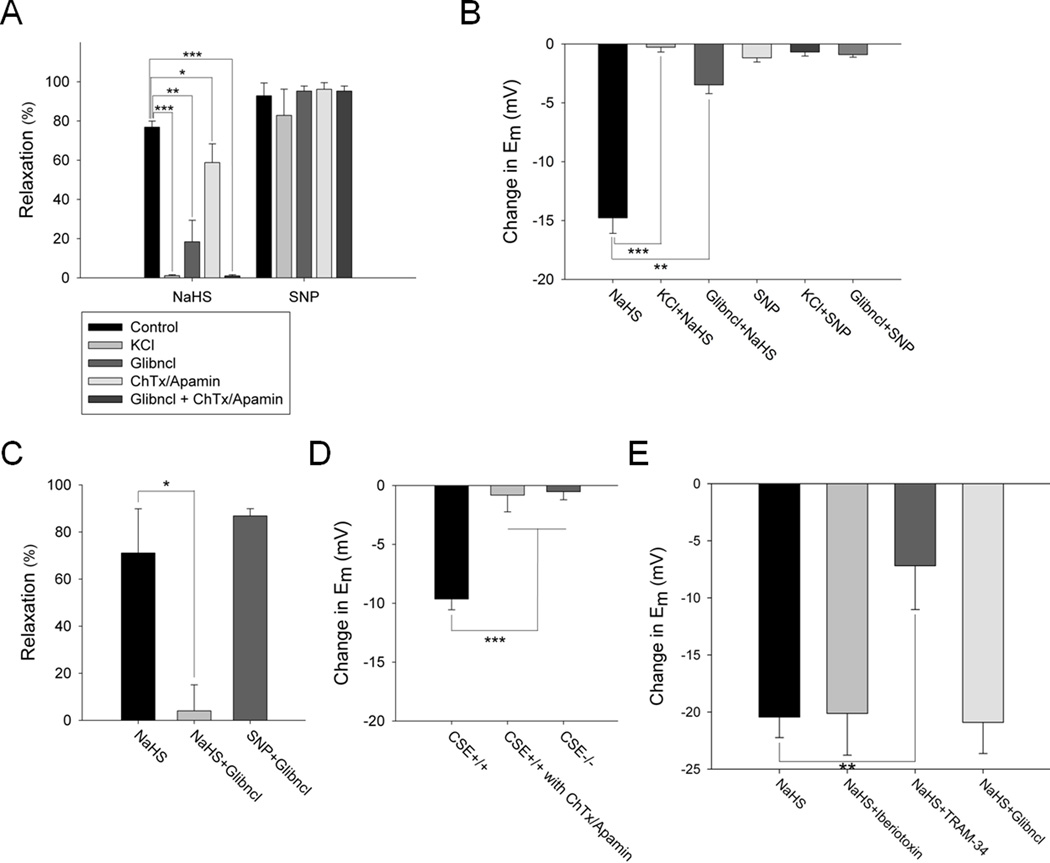
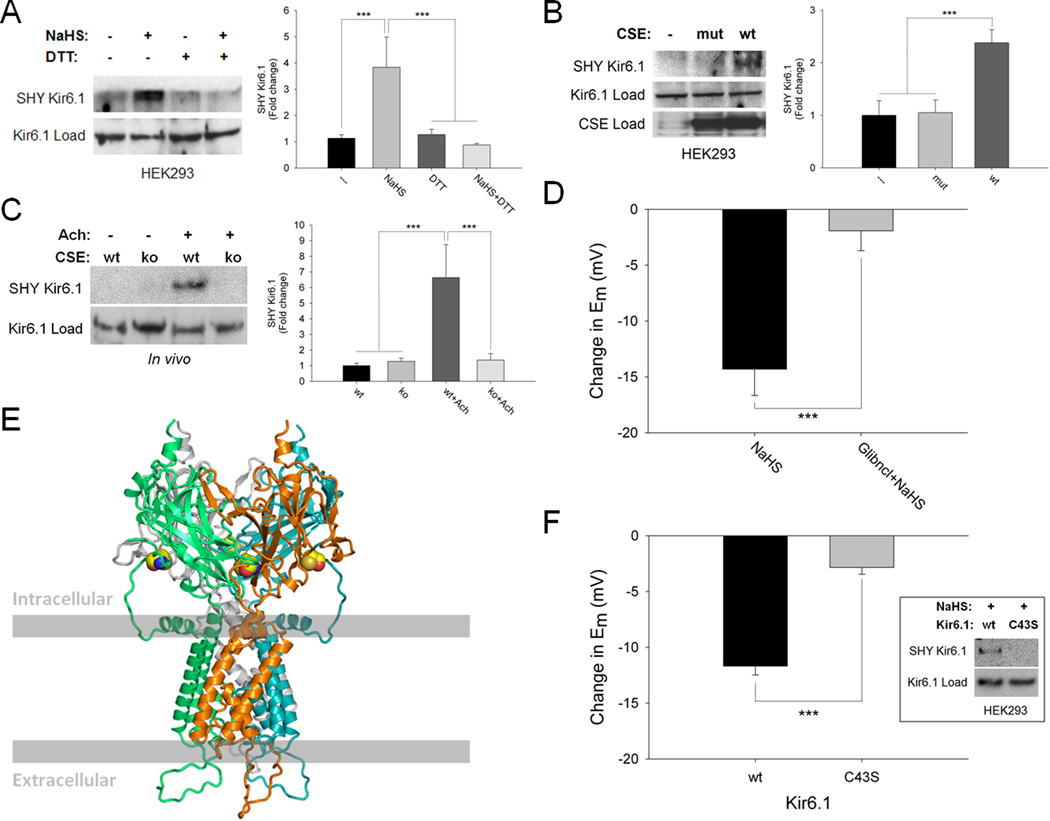
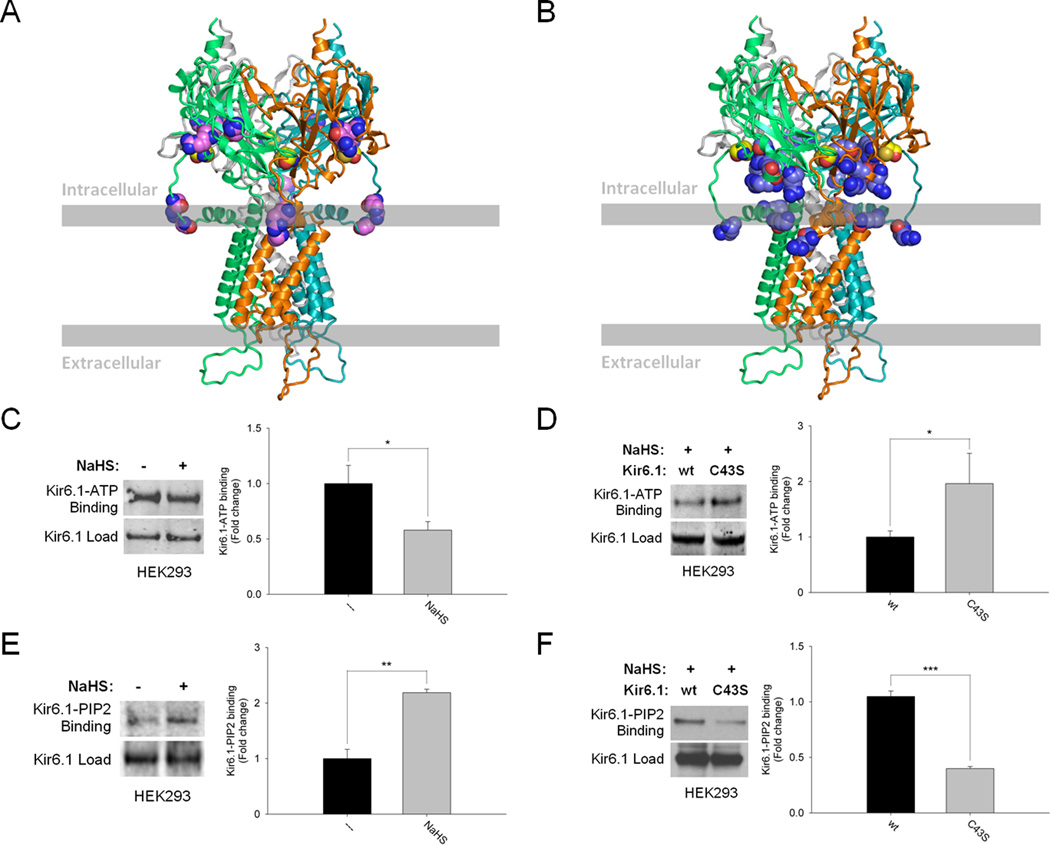
Methods and results: We now show that H(2)S is a major EDHF because in blood vessels of CSE-deleted mice, hyperpolarization is virtually abolished. H(2)S acts by covalently modifying (sulfhydrating) the ATP-sensitive potassium channel, as mutating the site of sulfhydration prevents H(2)S-elicited hyperpolarization. The endothelial intermediate conductance (IK(Ca)) and small conductance (SK(Ca)) potassium channels mediate in part the effects of H(2)S, as selective IK(Ca) and SK(Ca) channel inhibitors, charybdotoxin and apamin, inhibit glibenclamide-insensitive, H(2)S-induced vasorelaxation.
Conclusions: H(2)S is a major EDHF that causes vascular endothelial and smooth muscle cell hyperpolarization and vasorelaxation by activating the ATP-sensitive, intermediate conductance and small conductance potassium channels through cysteine S-sulfhydration. Because EDHF activity is a principal determinant of vasorelaxation in numerous vascular beds, drugs influencing H(2)S biosynthesis offer therapeutic potential.
Figures
Comment in
-
Hydrogen sulfide as an endothelium-derived hyperpolarizing factor in rodent mesenteric arteries.Circ Res. 2012 Jan 6;110(1):e13-4; author reply e15-6. doi: 10.1161/CIRCRESAHA.111.259309. Circ Res. 2012. PMID: 22223214 No abstract available.
References
-
- Murad F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med. 2006 Nov 9;355(19):2003–2011. - PubMed
-
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980 Nov 27;288(5789):373–376. - PubMed
-
- Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarizations: past beliefs and present facts. Ann Med. 2007;39(7):495–516. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
