

High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism
- PMID: 21980284
- PMCID: PMC3182942
- DOI: 10.1371/journal.ppat.1002251
High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism
Abstract
The pathways that comprise cellular metabolism are highly interconnected, and alterations in individual enzymes can have far-reaching effects. As a result, global profiling methods that measure gene expression are of limited value in predicting how the loss of an individual function will affect the cell. In this work, we employed a new method of global phenotypic profiling to directly define the genes required for the growth of Mycobacterium tuberculosis. A combination of high-density mutagenesis and deep-sequencing was used to characterize the composition of complex mutant libraries exposed to different conditions. This allowed the unambiguous identification of the genes that are essential for Mtb to grow in vitro, and proved to be a significant improvement over previous approaches. To further explore functions that are required for persistence in the host, we defined the pathways necessary for the utilization of cholesterol, a critical carbon source during infection. Few of the genes we identified had previously been implicated in this adaptation by transcriptional profiling, and only a fraction were encoded in the chromosomal region known to encode sterol catabolic functions. These genes comprise an unexpectedly large percentage of those previously shown to be required for bacterial growth in mouse tissue. Thus, this single nutritional change accounts for a significant fraction of the adaption to the host. This work provides the most comprehensive genetic characterization of a sterol catabolic pathway to date, suggests putative roles for uncharacterized virulence genes, and precisely maps genes encoding potential drug targets.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
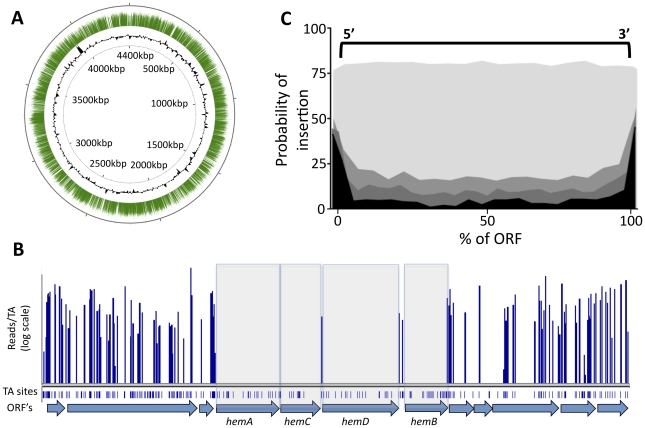
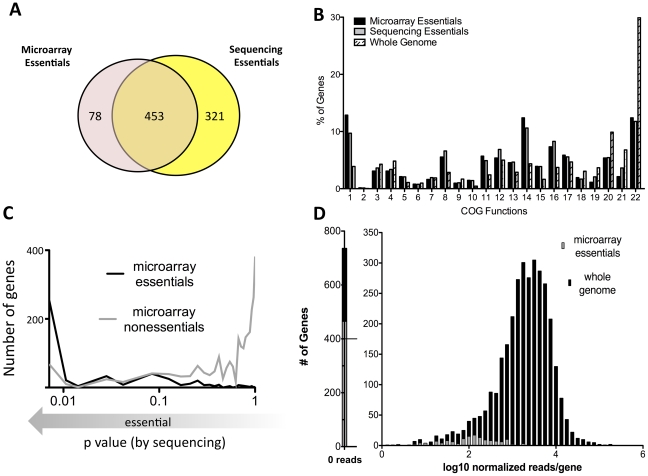
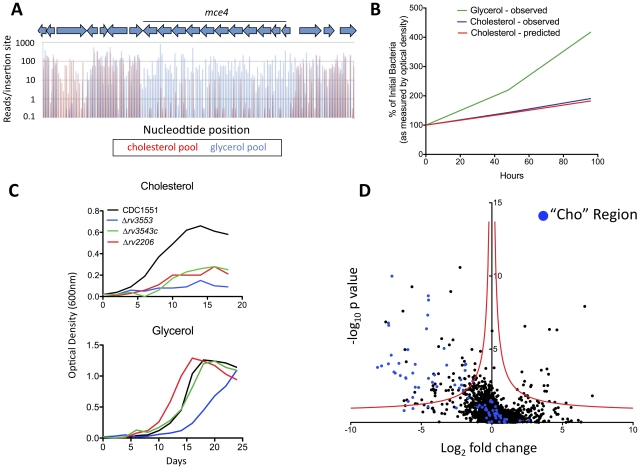
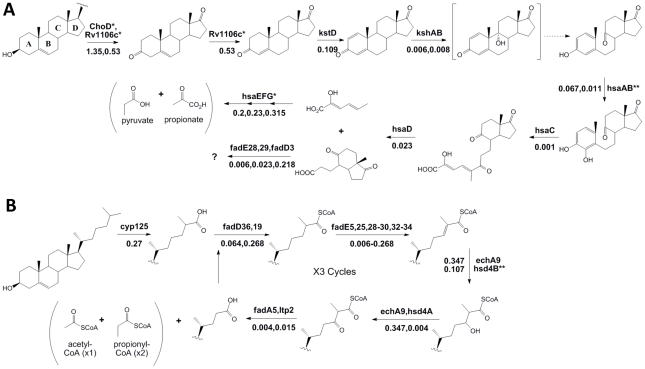
References
-
- World Health Organization. Global Tuberculosis Control: Surveillance P, Financing. WHO Report. 2010 http://www.who.int/tb/publications/global_report/2010/en/index.html.
-
- Badarinarayana V, Estep PW, Shendure J, Edwards J, Tavazoie S, et al. Selection analyses of insertional mutants using subgenic-resolution arrays. Nat Biotechnol. 2001;19:1060–1065. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
