

Practical tools to implement massive parallel pyrosequencing of PCR products in next generation molecular diagnostics
- PMID: 21980484
- PMCID: PMC3184136
- DOI: 10.1371/journal.pone.0025531
Practical tools to implement massive parallel pyrosequencing of PCR products in next generation molecular diagnostics
Abstract
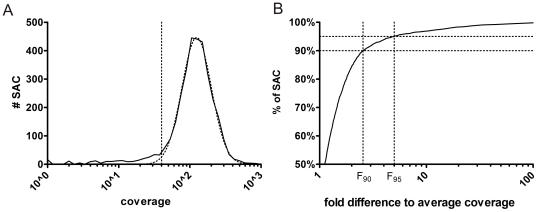
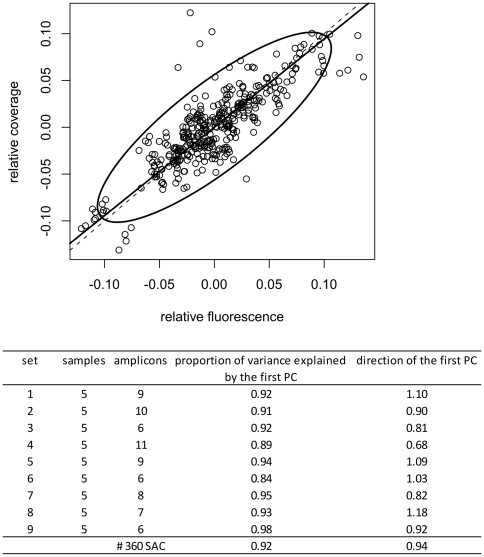
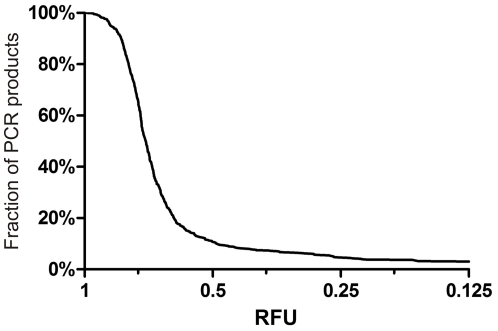
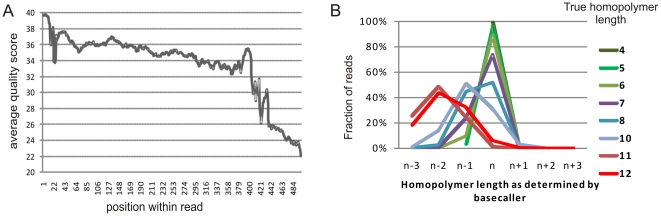
Despite improvements in terms of sequence quality and price per basepair, Sanger sequencing remains restricted to screening of individual disease genes. The development of massively parallel sequencing (MPS) technologies heralded an era in which molecular diagnostics for multigenic disorders becomes reality. Here, we outline different PCR amplification based strategies for the screening of a multitude of genes in a patient cohort. We performed a thorough evaluation in terms of set-up, coverage and sequencing variants on the data of 10 GS-FLX experiments (over 200 patients). Crucially, we determined the actual coverage that is required for reliable diagnostic results using MPS, and provide a tool to calculate the number of patients that can be screened in a single run. Finally, we provide an overview of factors contributing to false negative or false positive mutation calls and suggest ways to maximize sensitivity and specificity, both important in a routine setting. By describing practical strategies for screening of multigenic disorders in a multitude of samples and providing answers to questions about minimum required coverage, the number of patients that can be screened in a single run and the factors that may affect sensitivity and specificity we hope to facilitate the implementation of MPS technology in molecular diagnostics.
Conflict of interest statement
Figures
References
-
- Chou LS, Lyon E, Wittwer CT. A comparison of high-resolution melting analysis with denaturing high-performance liquid chromatography for mutation scanning: cystic fibrosis transmembrane conductance regulator gene as a model. American journal of clinical pathology. 2005;124:330–338. - PubMed
-
- Wittwer CT. High-resolution DNA melting analysis: advancements and limitations. Human mutation. 2009;30:857–859. - PubMed
-
- De Leeneer K, Coene I, Poppe B, De Paepe A, Claes K. Rapid and sensitive detection of BRCA1/2 mutations in a diagnostic setting: comparison of two high-resolution melting platforms. Clinical chemistry. 2008;54:982–989. - PubMed
-
- Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. Journal of molecular biology. 1975;94:441–448. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
