

Allosteric function and dysfunction of the prion protein
- PMID: 21984610
- PMCID: PMC11114699
- DOI: 10.1007/s00018-011-0847-7
Allosteric function and dysfunction of the prion protein
Abstract
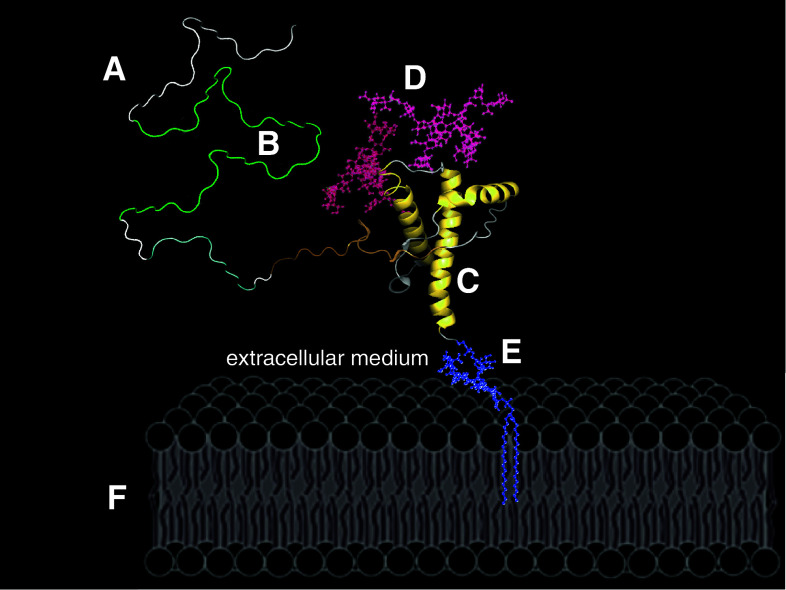
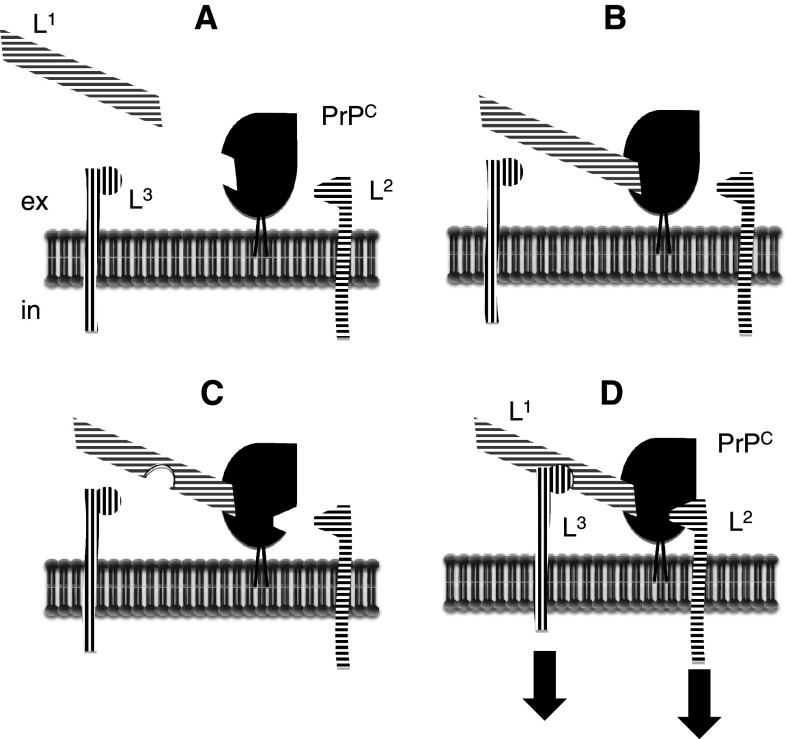
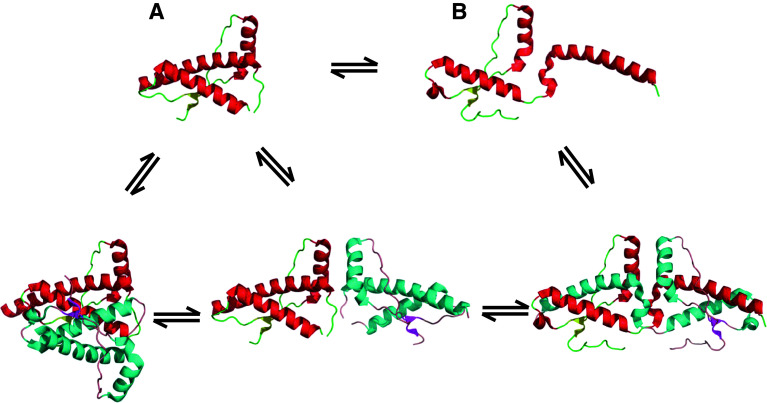
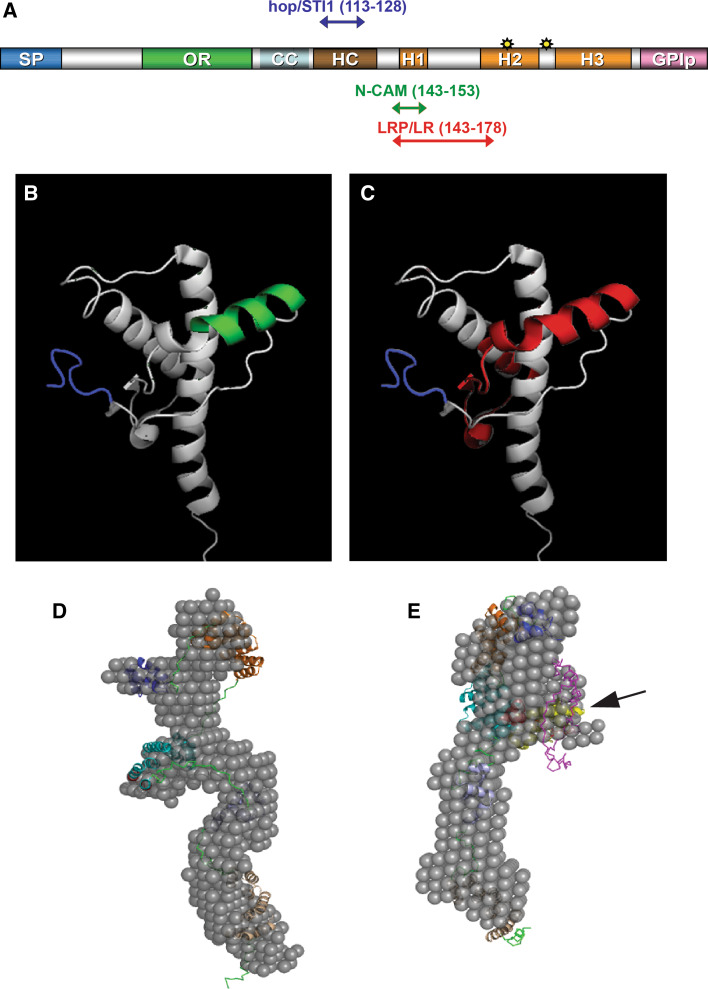
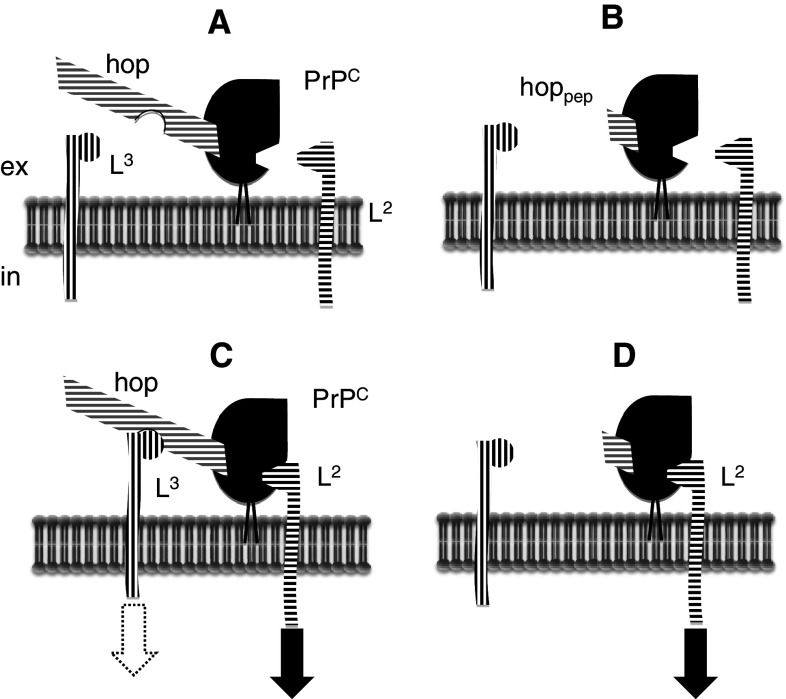
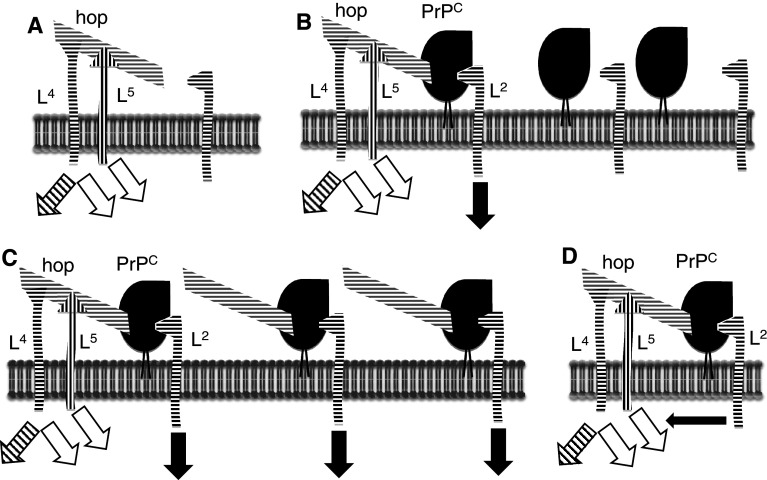
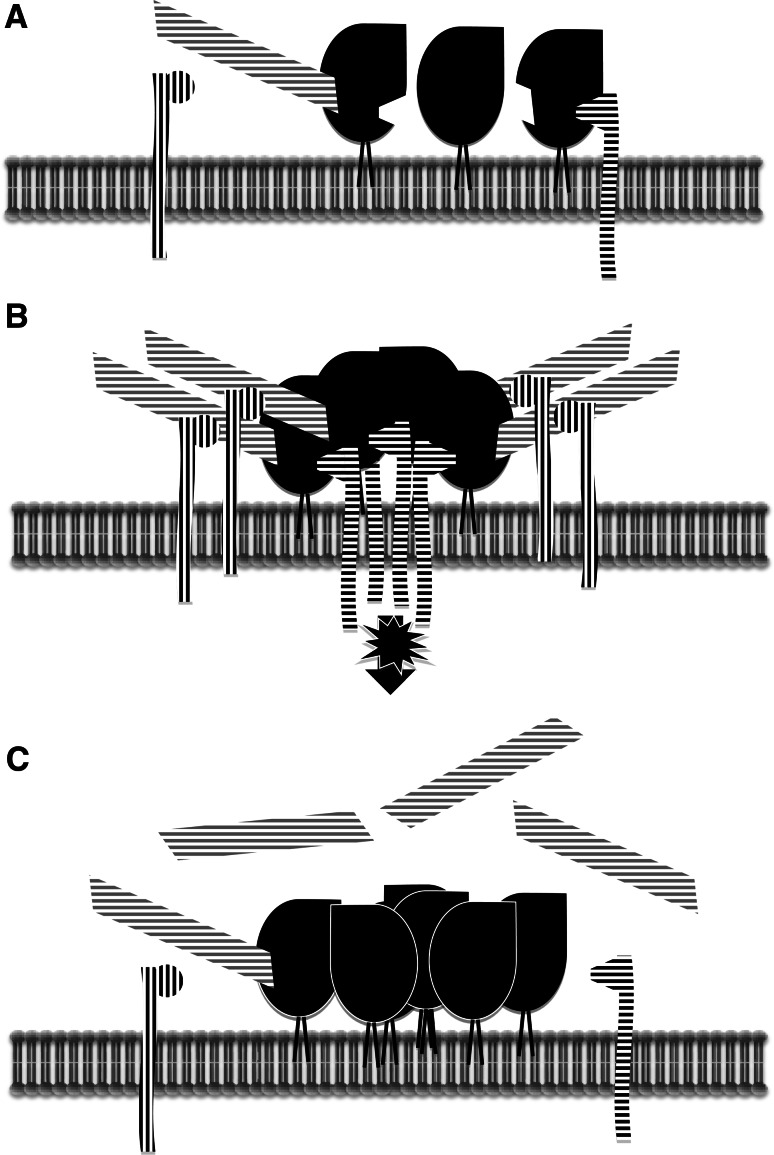
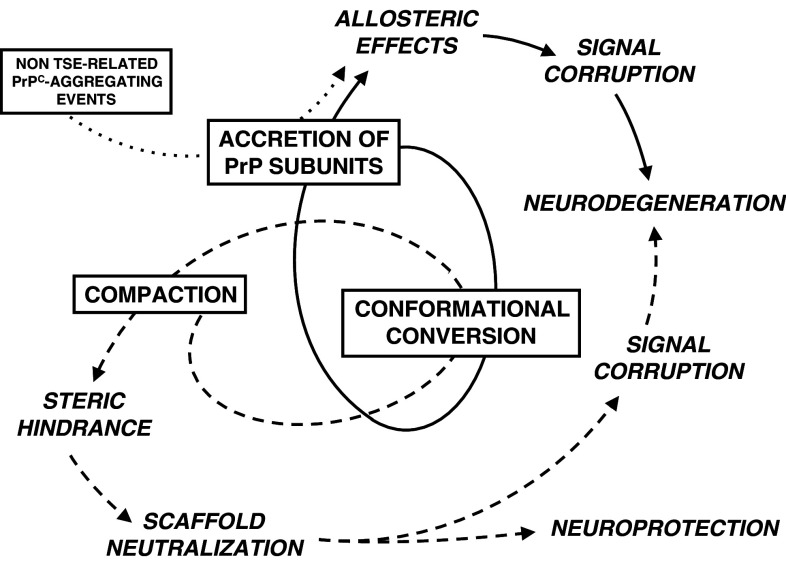
Transmissible spongiform encephalopathies (TSEs) are neurodegenerative diseases associated with progressive oligo- and multimerization of the prion protein (PrP(C)), its conformational conversion, aggregation and precipitation. We recently proposed that PrP(C) serves as a cell surface scaffold protein for a variety of signaling modules, the effects of which translate into wide-range functional consequences. Here we review evidence for allosteric functions of PrP(C), which constitute a common property of scaffold proteins. The available data suggest that allosteric effects among PrP(C) and its partners are involved in the assembly of multi-component signaling modules at the cell surface, impose upon both physiological and pathological conformational responses of PrP(C), and that allosteric dysfunction of PrP(C) has the potential to entail progressive signal corruption. These properties may be germane both to physiological roles of PrP(C), as well as to the pathogenesis of the TSEs and other degenerative/non-communicable diseases.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
