

High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability
- PMID: 21984976
- PMCID: PMC3187555
- DOI: 10.1158/2159-8290.CD-11-0039
High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability
Erratum in
- Cancer Discov. 2012 Aug;2(8):750-1
-
Correction: High Frequency of PIK3R1 and PIK3R2 Mutations in Endometrial Cancer Elucidates a Novel Mechanism for Regulation of PTEN Protein Stability.Cancer Discov. 2021 Oct;11(10):2658. doi: 10.1158/2159-8290.CD-21-1070. Cancer Discov. 2021. PMID: 34598952 No abstract available.
Abstract
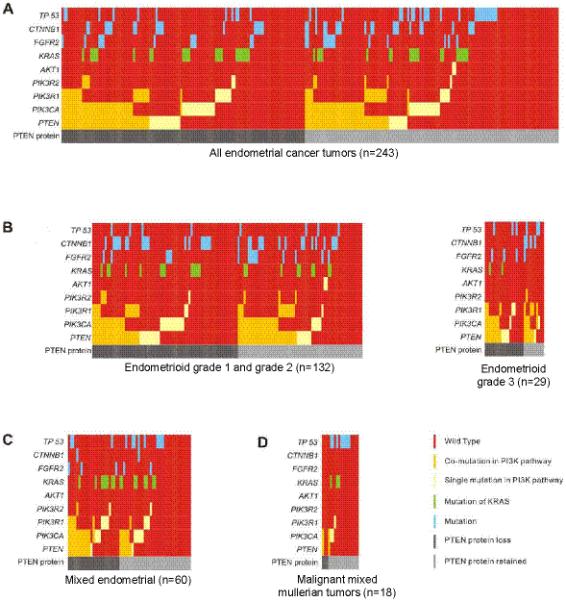
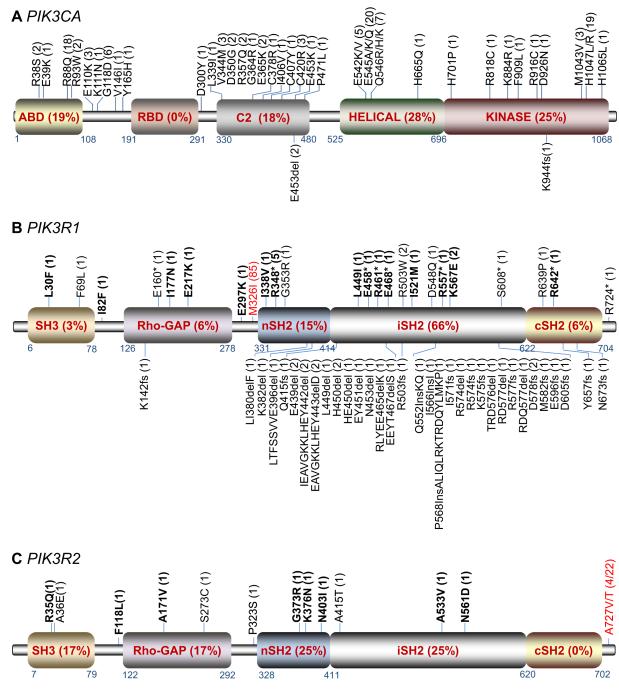
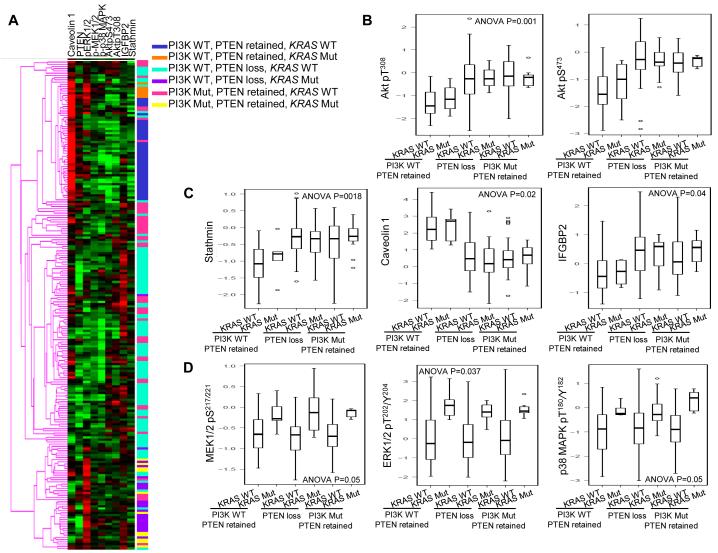
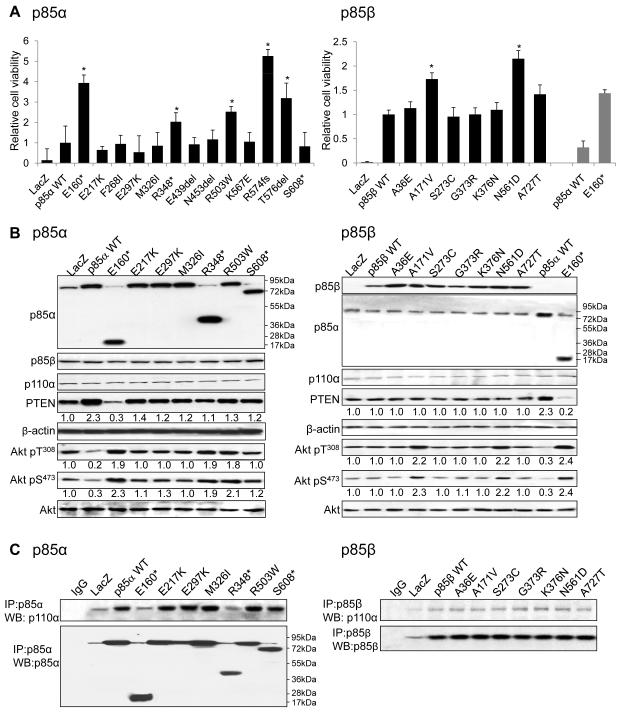
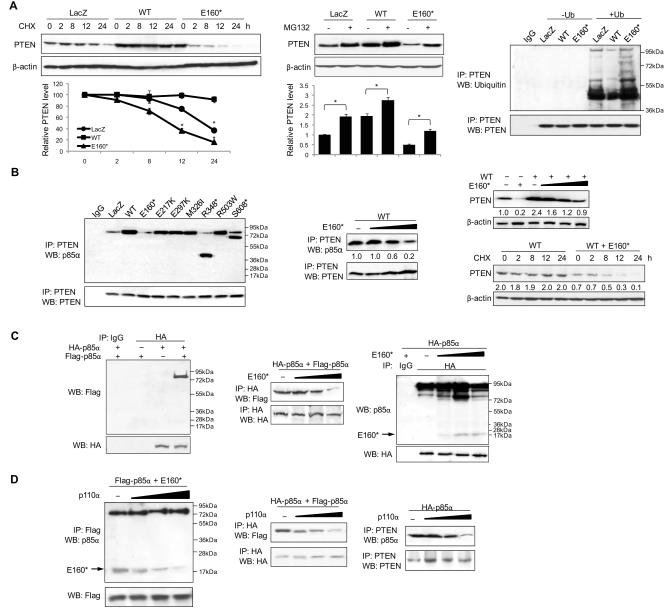
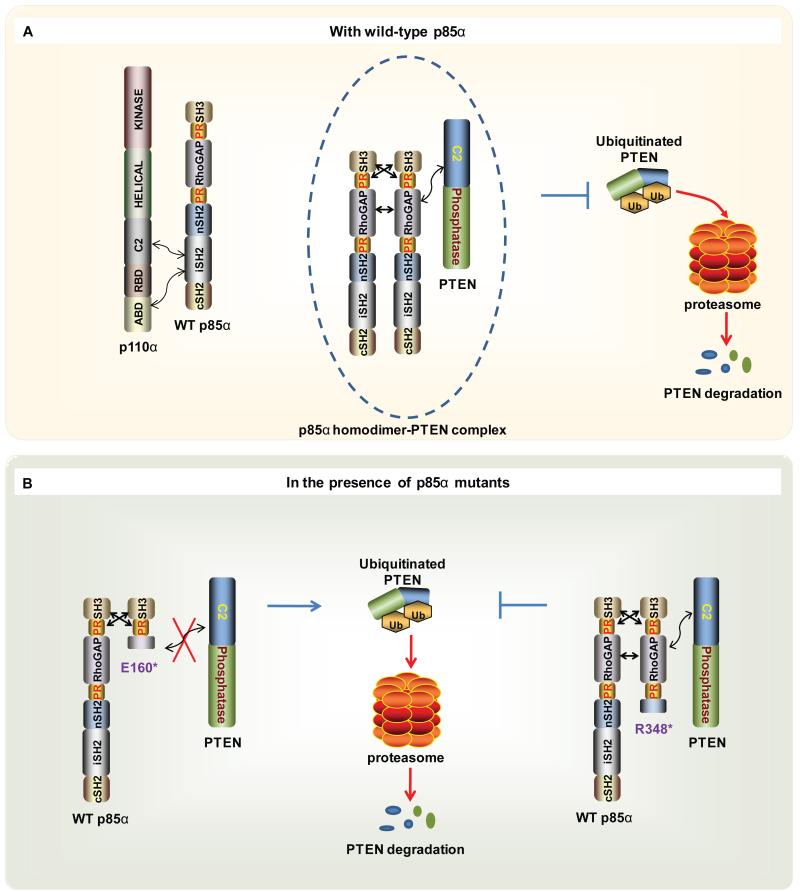
We demonstrate that phosphatidylinositol 3-kinase (PI3K) pathway aberrations occur in >80% of endometrioid endometrial cancers, with coordinate mutations of multiple PI3K pathway members being more common than predicted by chance. PIK3R1 (p85α) mutations occur at a higher rate in endometrial cancer than in any other tumor lineage, and PIK3R2 (p85β), not previously demonstrated to be a cancer gene, is also frequently mutated. The dominant activation event in the PI3K pathway appears to be PTEN protein loss. However, in tumors with retained PTEN protein, PI3K pathway mutations phenocopy PTEN loss, resulting in pathway activation. KRAS mutations are common in endometrioid tumors activating independent events from PI3K pathway aberrations. Multiple PIK3R1 and PIK3R2 mutations demonstrate gain of function, including disruption of a novel mechanism of pathway regulation wherein p85α dimers bind and stabilize PTEN. Taken together, the PI3K pathway represents a critical driver of endometrial cancer pathogenesis and a novel therapeutic target.
Keywords: Endometrial Cancer; PIK3CA; PIK3R1; PIK3R2; PTEN.
Figures
Comment in
-
New routes to old places: PIK3R1 and PIK3R2 join PIK3CA and PTEN as endometrial cancer genes.Cancer Discov. 2011 Jul;1(2):106-7. doi: 10.1158/2159-8290.CD-11-0116. Cancer Discov. 2011. PMID: 22586352
References
-
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. - PubMed
-
- Mutter GL, Ince TA, Baak JP, Kust GA, Zhou XP, Eng C. Molecular identification of latent precancers in histologically normal endometrium. Cancer Res. 2001;61:4311–4. - PubMed
-
- Sun H, Enomoto T, Fujita M, Wada H, Yoshino K, Ozaki K, et al. Mutational analysis of the PTEN gene in endometrial carcinoma and hyperplasia. Am J Clin Pathol. 2001;115:32–8. - PubMed
-
- Kanamori Y, Kigawa J, Itamochi H, Shimada M, Takahashi M, Kamazawa S, et al. Correlation between loss of PTEN expression and Akt phosphorylation in endometrial carcinoma. Clin Cancer Res. 2001;7:892–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
