

Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation
- PMID: 21987724
- PMCID: PMC3228869
- DOI: 10.1158/0008-5472.CAN-11-1514
Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation
Abstract
Glioblastoma multiforme (GBM) is characterized by overexpression of epidermal growth factor receptor (EGFR) and loss of the tumor suppressors Ink4a/Arf. Efforts at modeling GBM using wild-type EGFR in mice have proven unsuccessful. Here, we present a unique mouse model of wild-type EGFR-driven gliomagenesis. We used a combination of somatic conditional overexpression and ligand-mediated chronic activation of EGFR in cooperation with Ink4a/Arf loss in the central nervous system of adult mice to generate tumors with the histopathologic and molecular characteristics of human GBMs. Sustained, ligand-mediated activation of EGFR was necessary for gliomagenesis, functionally substantiating the clinical observation that EGFR-positive GBMs from patients express EGFR ligands. To gain a better understanding of the clinically disappointing EGFR-targeted therapies for GBM, we investigated the molecular responses to EGFR tyrosine kinase inhibitor (TKI) treatment in this model. Gefitinib treatment of primary GBM cells resulted in a robust apoptotic response, partially conveyed by mitogen-activated protein kinase (MAPK) signaling attenuation and accompanied by BIM(EL) expression. In human GBMs, loss-of-function mutations in the tumor suppressor PTEN are a common occurrence. Elimination of PTEN expression in GBM cells posttumor formation did not confer resistance to TKI treatment, showing that PTEN status in our model is not predictive. Together, these findings offer important mechanistic insights into the genetic determinants of EGFR gliomagenesis and sensitivity to TKIs and provide a robust discovery platform to better understand the molecular events that are associated with predictive markers of TKI therapy.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures
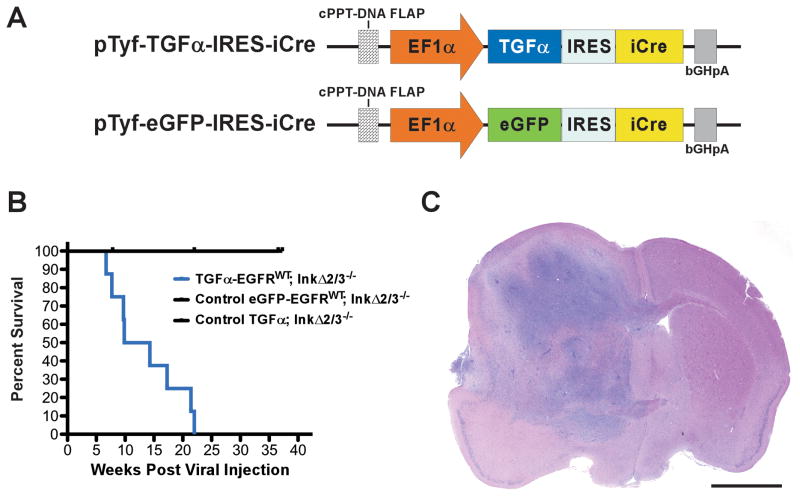
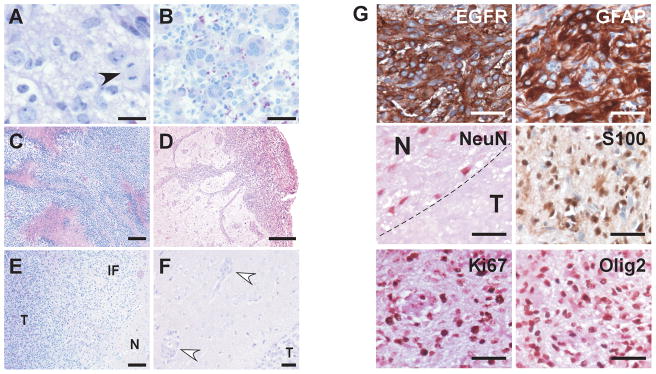
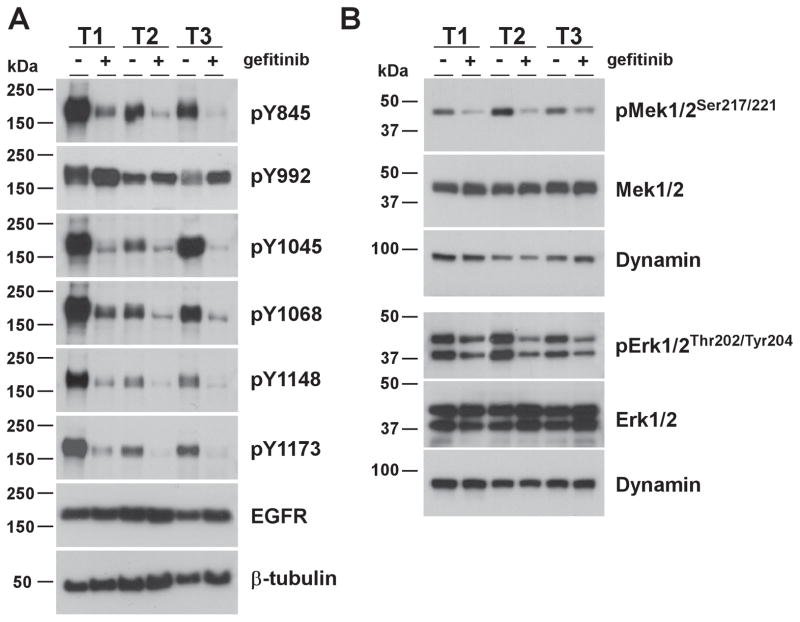
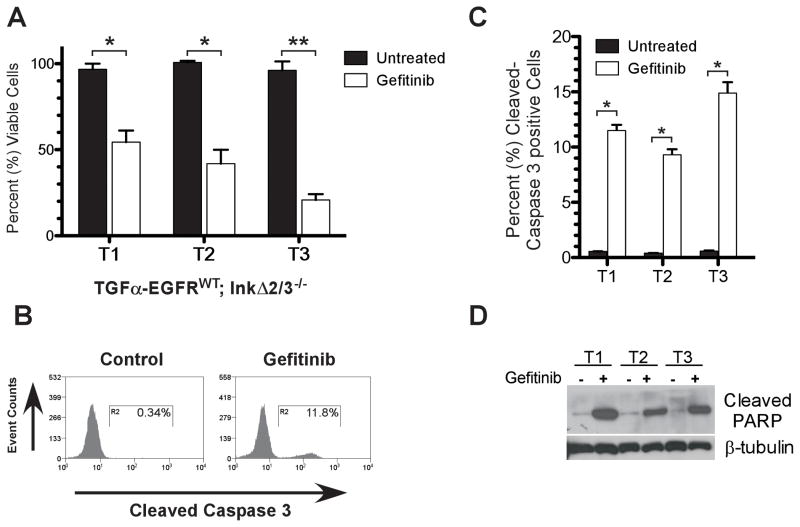
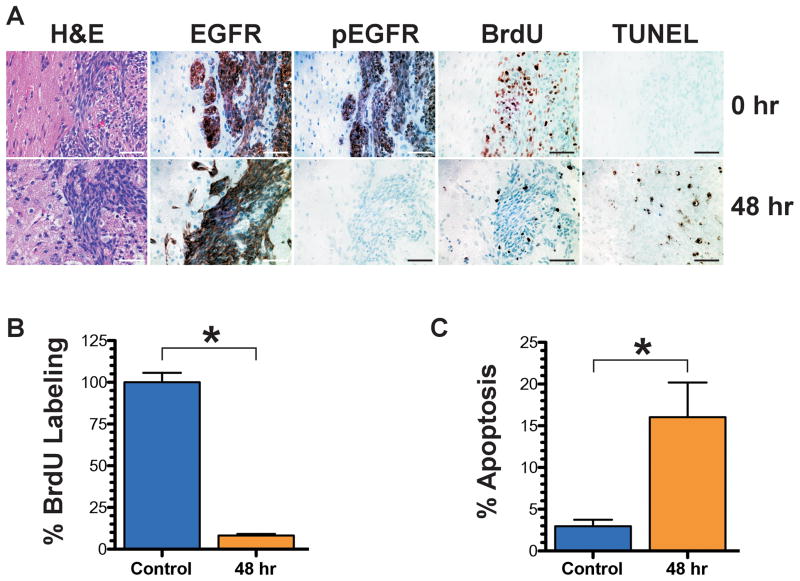
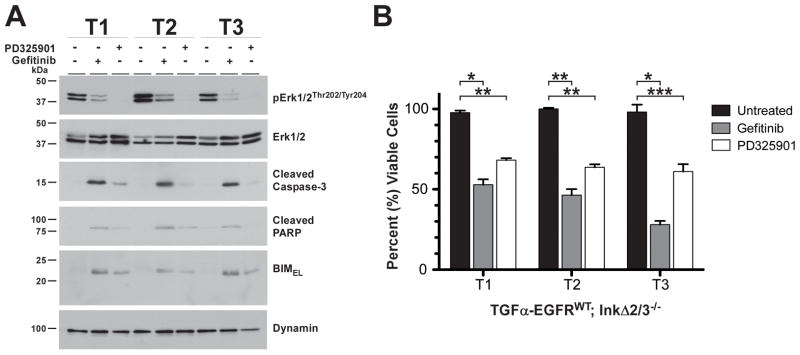
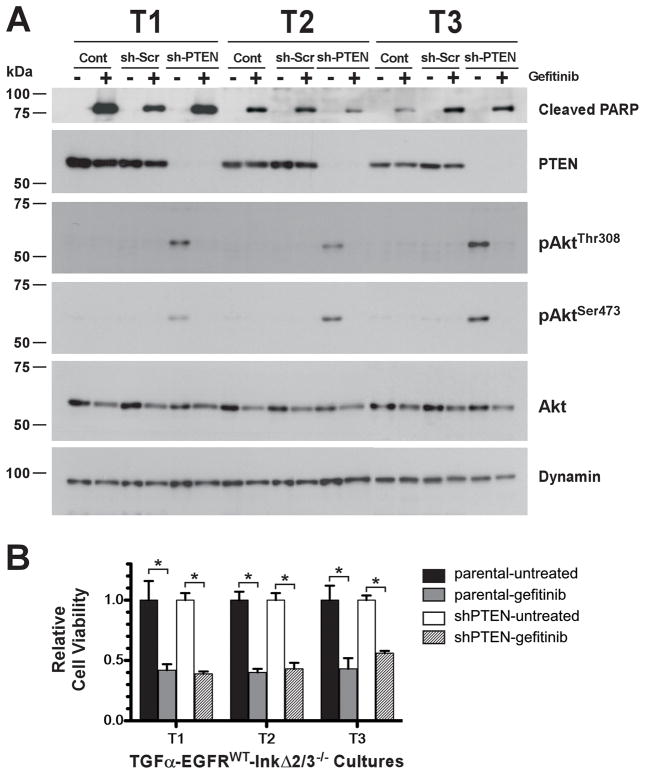
References
-
- Brennan C. Genomic profiles of glioma. Curr Neurol Neurosci Rep. 2011;11:291–7. - PubMed
-
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. - PubMed
-
- Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Sci Signal. 2009;2:re6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
