

Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins
- PMID: 21991322
- PMCID: PMC3184992
- DOI: 10.1371/journal.pone.0025590
Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins
Abstract
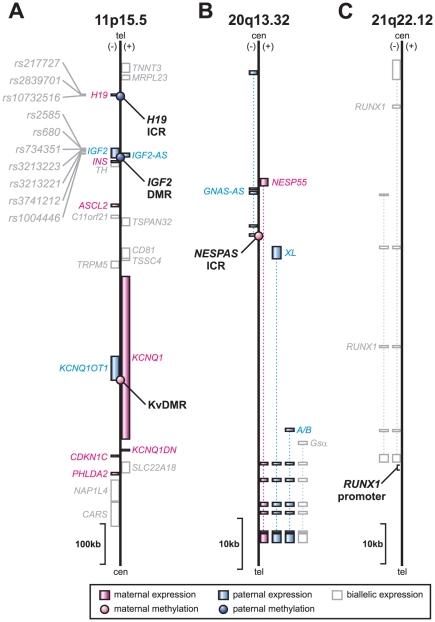
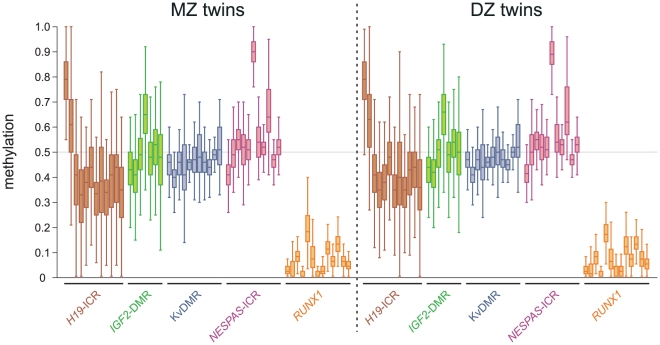
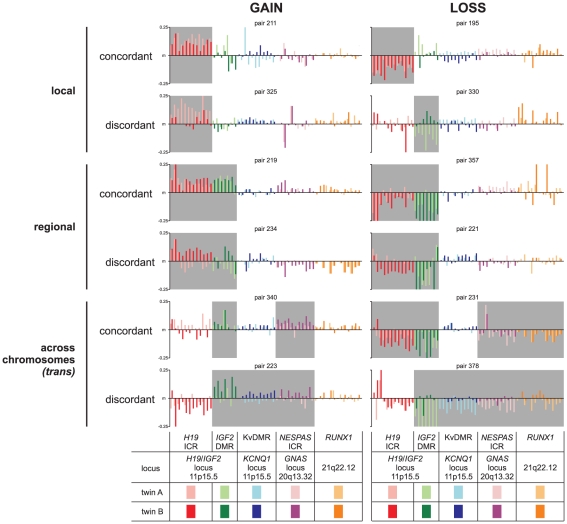
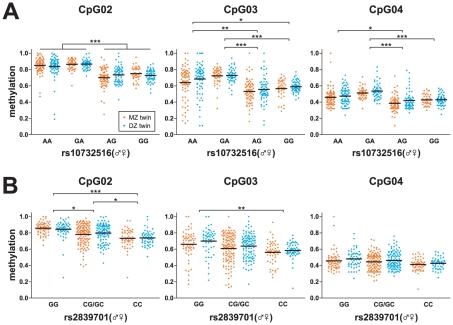
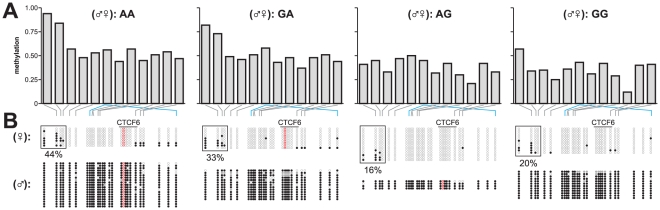
One of the best studied read-outs of epigenetic change is the differential expression of imprinted genes, controlled by differential methylation of imprinted control regions (ICRs). To address the impact of genotype on the epigenome, we performed a detailed study in 128 pairs of monozygotic (MZ) and 128 pairs of dizygotic (DZ) twins, interrogating the DNA methylation status of the ICRs of IGF2, H19, KCNQ1, GNAS and the non-imprinted gene RUNX1. While we found a similar overall pattern of methylation between MZ and DZ twins, we also observed a high degree of variability in individual CpG methylation levels, notably at the H19/IGF2 loci. A degree of methylation plasticity independent of the genome sequence was observed, with both local and regional CpG methylation changes, discordant between MZ and DZ individual pairs. However, concordant gains or losses of methylation, within individual twin pairs were more common in MZ than DZ twin pairs, indicating that de novo and/or maintenance methylation is influenced by the underlying DNA sequence. Specifically, for the first time we showed that the rs10732516 [A] polymorphism, located in a critical CTCF binding site in the H19 ICR locus, is strongly associated with increased hypermethylation of specific CpG sites in the maternal H19 allele. Together, our results highlight the impact of the genome on the epigenome and demonstrate that while DNA methylation states are tightly maintained between genetically identical and related individuals, there remains considerable epigenetic variation that may contribute to disease susceptibility.
Conflict of interest statement
Figures



References
-
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. - PubMed
-
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. - PubMed
-
- Bartolomei MS, Zemel S, Tilghman SM. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. - PubMed
-
- DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849–859. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
