

Improving the prediction of disease-related variants using protein three-dimensional structure
- PMID: 21992054
- PMCID: PMC3194195
- DOI: 10.1186/1471-2105-12-S4-S3
Improving the prediction of disease-related variants using protein three-dimensional structure
Abstract
Background: Single Nucleotide Polymorphisms (SNPs) are an important source of human genome variability. Non-synonymous SNPs occurring in coding regions result in single amino acid polymorphisms (SAPs) that may affect protein function and lead to pathology. Several methods attempt to estimate the impact of SAPs using different sources of information. Although sequence-based predictors have shown good performance, the quality of these predictions can be further improved by introducing new features derived from three-dimensional protein structures.
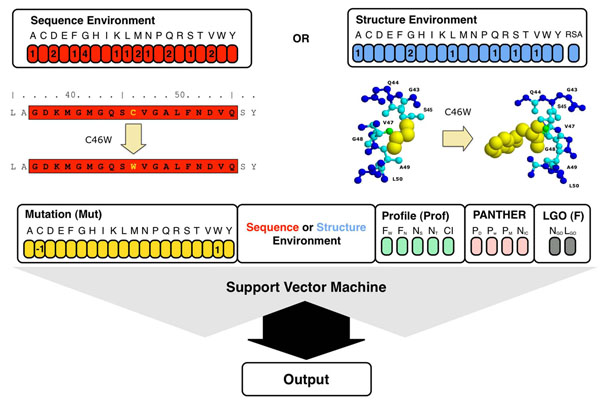
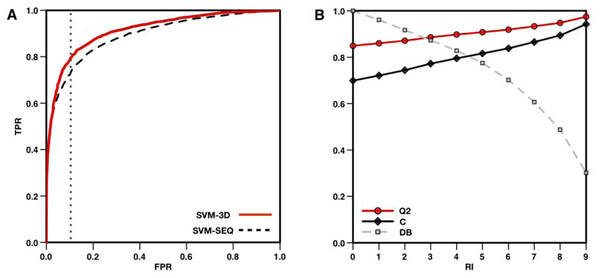
Results: In this paper, we present a structure-based machine learning approach for predicting disease-related SAPs. We have trained a Support Vector Machine (SVM) on a set of 3,342 disease-related mutations and 1,644 neutral polymorphisms from 784 protein chains. We use SVM input features derived from the protein's sequence, structure, and function. After dataset balancing, the structure-based method (SVM-3D) reaches an overall accuracy of 85%, a correlation coefficient of 0.70, and an area under the receiving operating characteristic curve (AUC) of 0.92. When compared with a similar sequence-based predictor, SVM-3D results in an increase of the overall accuracy and AUC by 3%, and correlation coefficient by 0.06. The robustness of this improvement has been tested on different datasets and in all the cases SVM-3D performs better than previously developed methods even when compared with PolyPhen2, which explicitly considers in input protein structure information.
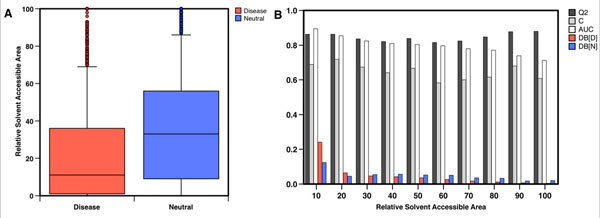
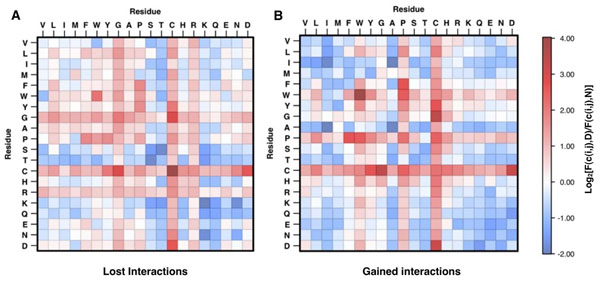
Conclusion: This work demonstrates that structural information can increase the accuracy of disease-related SAPs identification. Our results also quantify the magnitude of improvement on a large dataset. This improvement is in agreement with previously observed results, where structure information enhanced the prediction of protein stability changes upon mutation. Although the structural information contained in the Protein Data Bank is limiting the application and the performance of our structure-based method, we expect that SVM-3D will result in higher accuracy when more structural date become available.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
