

Review
doi: 10.1038/nrc3106.
RAS oncogenes: weaving a tumorigenic web
Affiliations
- PMID: 21993244
- PMCID: PMC3632399
- DOI: 10.1038/nrc3106
Item in Clipboard
Review
RAS oncogenes: weaving a tumorigenic web
Nat Rev Cancer.
.
Abstract
RAS proteins are essential components of signalling pathways that emanate from cell surface receptors. Oncogenic activation of these proteins owing to missense mutations is frequently detected in several types of cancer. A wealth of biochemical and genetic studies indicates that RAS proteins control a complex molecular circuitry that consists of a wide array of interconnecting pathways. In this Review, we describe how RAS oncogenes exploit their extensive signalling reach to affect multiple cellular processes that drive tumorigenesis.
Conflict of interest statement
Figures
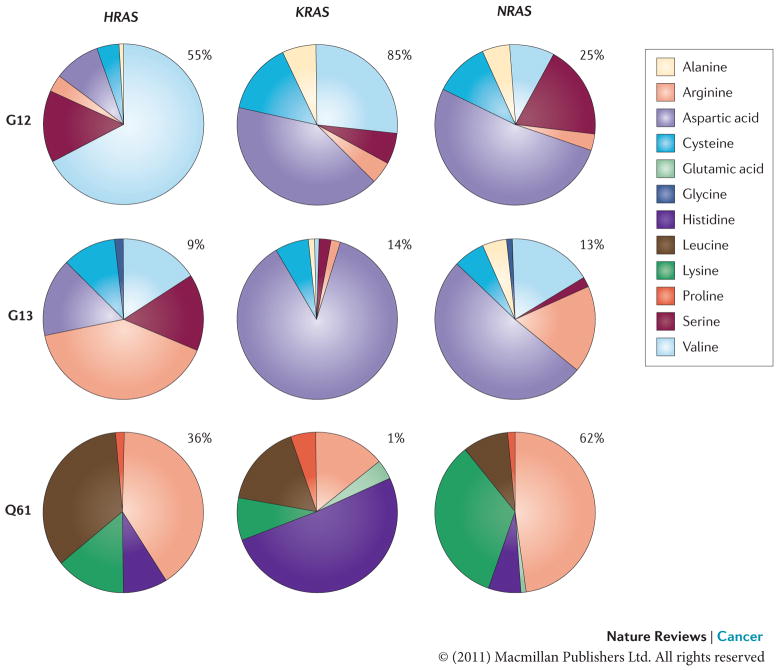
The frequency of mutational substitution at G12, G13 or Q61 for a particular amino acid has been represented using pie charts. Percentages indicate the frequency with which a given residue is mutated within a particular isoform. Primary data source is the COSMIC database (see Further information).
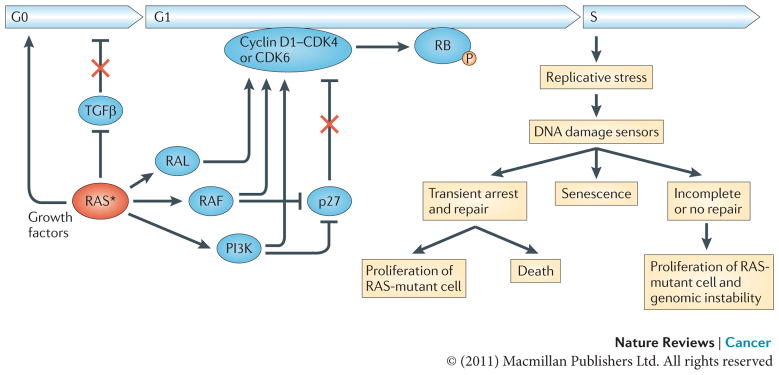
Oncogenic RAS establishes independence from extracellular growth factors and growth inhibitors, thereby promoting exit from the G0 phase of the cell cycle, progression through G1 and entry into S phase. RAS induces the transcriptional upregulation of growth factors and interferes with transforming growth factor-β (TGFβ) signalling through inhibition of TGFβ receptor expression or downstream signalling by downregulating the expression of SMAD3, as well as the nuclear accumulation of SMAD2 and SMAD3. RAS also upregulates the levels of cyclin D1 and suppresses the cyclin-dependent kinase inhibitor (CDKI) p27. The newly synthesized cyclin D1 associates with and activates the cyclin-dependent kinases CDK4 and CDK6, leading to the phosphorylation of RB and the subsequent dissolution of the RB–E2F transcription factor complexes. Once released, E2F transcription factors transactivate several genes that are required for cell cycle progression, including cyclin E (CCNE) and cyclin A (CCNA) that induce transition through the G1/S checkpoint (not shown). Hyperproliferative cues from activation of the RAS oncogene can result in replicative stress leading to DNA damage. In response to DNA damage cells can activate the DNA damage checkpoints to transiently arrest and restore the integrity of the genome, enter a state of irreversible arrest (senescence) or undergo apoptosis. Inaccurate repair of DNA damage can lead to mutations and chromosome aberrations, thereby contributing to tumorigenesis. The asterisk represents the mutational activation of RAS. P, phosphorylation.
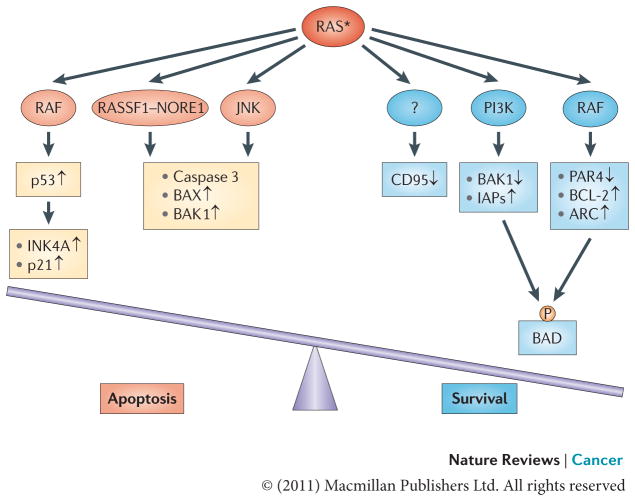
Oncogenic RAS may have both pro-apoptotic and anti-apoptotic functions depending on the status of RAS effector pathways and the apoptotic machinery. In many cases oncogenic RAS signalling through the RAF pathway engages an apoptotic response that is mediated by p53. Also, signalling through the RAS effectors RASSF1, NORE1, mammalian STE20-like protein kinase 1 (MST1) and JUN N-terminal kinase (JNK) can lead to apoptotic death via the activation of caspase 3 and the pro-apoptotic proteins BCL-2-associated X protein (BAX) and BCL-2-homologous antagonist/killer 1 (BAK1). Acquisition of a tumorigenic phenotype is marked by the suppression of such mediators of RAS-induced apoptosis. In this context, the anti-apoptotic activity of RAS takes a stronghold. The anti-apoptotic function of oncogenic RAS is mediated by several effector pathways, including the RAS–PI3K effector pathway, which regulates the levels of pro-apoptotic protein BAK1 and inhibitors of apoptosis (IAPs), and the RAS–RAF pathway, which downregulates the pro-apoptotic transcriptional repressor prostate apoptosis response 4 (PAR4) while upregulating the anti-apoptotic proteins BCL-2 and apoptosis repressor with caspase recruitment domain (ARC). Both pathways have been implicated in phosphorylating and inactivating the pro-apoptotic protein BCL-2-associated agonist of cell death (BAD). The mechanism through which RAS induces the epigenetic silencing of the pro-apoptotic CD95 gene remains to be uncovered. The asterisk represents the mutational activation of RAS protein. P, phosphorylation.
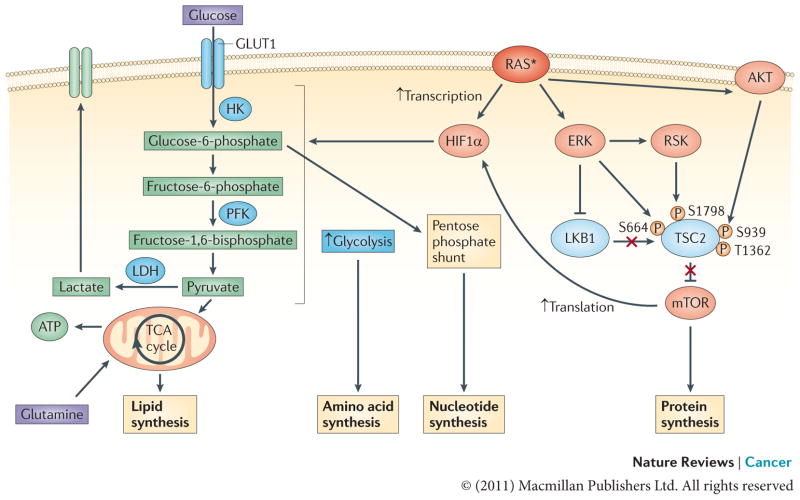
ERK and PI3K signalling downstream of oncogenic RAS converge to activate mTOR by inhibiting its negative regulators tuberin (TSC2) and liver kinase B1 (LKB1)–AMP-activated protein kinase (AMPK). TSC2 can be directly phosphorylated by both ERK and ERK-activated ribosomal protein S6 kinase (RSK) on S664 and S1798, respectively, as well as by AKT (on S939 and T1362), and, likewise, RAF–ERK1 or RAF–ERK2 signalling disrupts the LKB1–AMPK checkpoint–. This leads to mTOR–eukaryotic translation initiation factor 4 (eIF4)-dependent translation of hypoxia-inducible factor 1α (HIF1α). Activated RAS can also result in the transcriptional upregulation of HIF1A. Increased levels of HIF1α augment multiple steps in glycolytic metabolism (shown in blue). The upregulation of hexokinase (HK) facilitates the conversion of glucose to glucose-6-phosphate, a glycolytic intermediate that is used in pentose phosphate pathway-dependent nucleotide synthesis. Higher levels of phosphofructokinase (PFK) lead to an enhanced glycolytic flux and the production of pyruvate, which, in conjunction with the oncogenic RAS-dependent increase in lactose dehydrogenase (LDH) levels, can allow glycolysis to persist by regenerating NAD+, a necessary cofactor for glycolytic reactions,,. In addition, some of the pyruvate can enter the tricarboxylic acid (TCA) cycle where its conversion to citrate generates intermediates that are necessary for the synthesis of fatty acids and non-essential amino acids. The asterisk represents the mutational activation of RAS. GLUT1, glucose transporter 1.
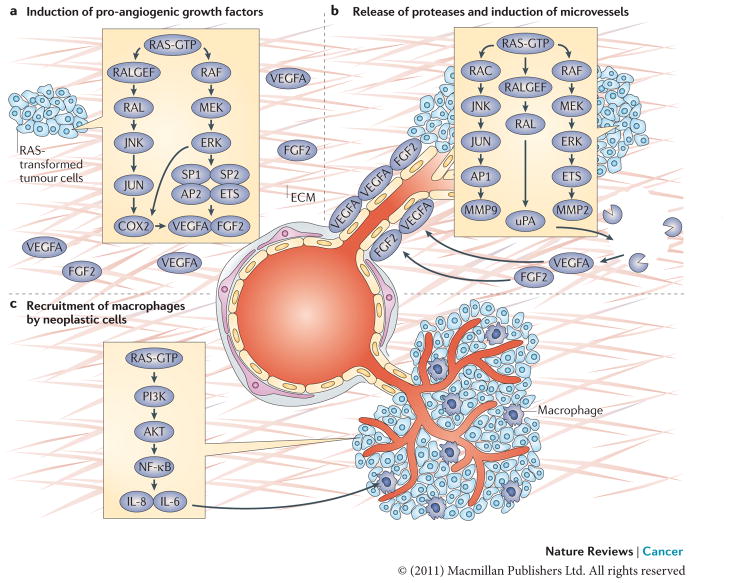
a. The induction of pro-angiogenic growth factors (vascular endothelial growth factor A (VEGFA) and fibroblast growth factor 2 (FGF2)) by RAS in neoplastic cells is shown. RAS enhances the transcription of VEGFA by recruiting transcription factors such as SP1, SP2, AP2 and ETS to the VEGFA promoter. RAS also increases the stability of VEGFA mRNA and augments its translation–. The RAS–JUN N-terminal kinase (JNK) signalling axis is responsible for upregulating the transcription of prostaglandin-endoperoxide synthase 2 (PTGS2), which encodes COX2, by activating JUN, a component of the AP1 transcription complex, whereas the RAS–ERK1 and ERK2 pathway contributes to COX2 expression through the phosphorylation of CCAAT/enhancer binding protein-β (C/EBPβ) and ETS transcription factors such as PEA3 (REFS –217). Expression of COX2, in turn, increases the levels of VEGFA produced by RAS-transformed cells. b. The release of proteases by neoplastic cells cleaves components of the extracellular matrix (ECM) and releases VEGFA and FGF2, which are trapped in the ECM. Expression of proteases urokinase-type plasminogen activator (uPA), matrix metalloproteinase 2 (MMP2) and MMP9 in RAS-transformed cells is increased by the combined effects of ETS transcription factors (activated by the RAF–ERK pathway) and JUN (activated by the RAC–JNK pathway) binding to the promoters of PEA3–AP1 sites, as well as enhanced translation of polysome-associated MMP9 mRNA,,. Stimulation of uPA expression is also dependent on RAS-mediated activation of RAL GTPase,. This induces neo-proliferation and sprouting of microvessels towards the tumour site. c. The recruitment of macrophages by neoplastic cells (through RAS-induced nuclear factor-κB (NF-κB)-dependent production of the cytokines interleukin-6 (IL-6) and IL-8) and subsequent promotion of endothelial proliferation and sprouting by newly recruited macrophages is shown.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. - PubMed
-
- Scheidig AJ, Burmester C, Goody RS. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure. 1999;7:1311–1324. - PubMed
-
- Scheffzek K, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. This article described the first three-dimensional structure of the RAS–RASGAP complex, providing insight into the mechanism of GTP hydrolysis and the structural basis for the oncogenicity of RAS mutants. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
