

Evidence of abundant stop codon readthrough in Drosophila and other metazoa
- PMID: 21994247
- PMCID: PMC3227100
- DOI: 10.1101/gr.119974.110
Evidence of abundant stop codon readthrough in Drosophila and other metazoa
Abstract
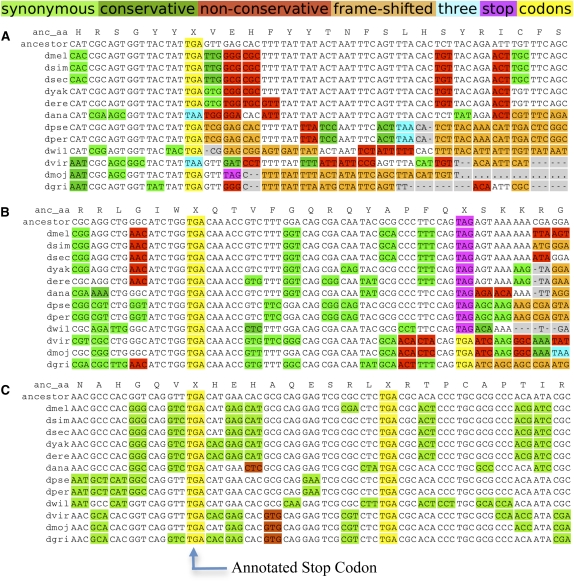
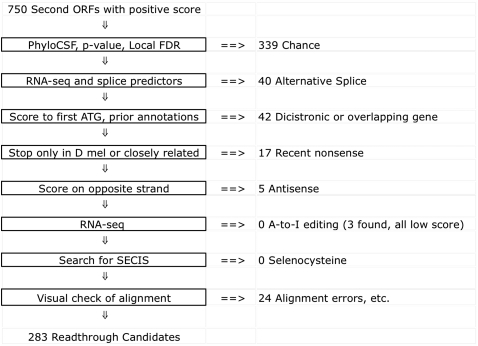
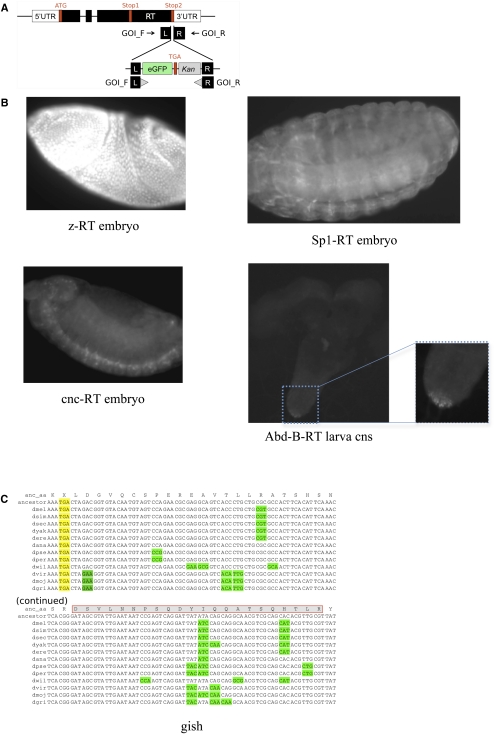
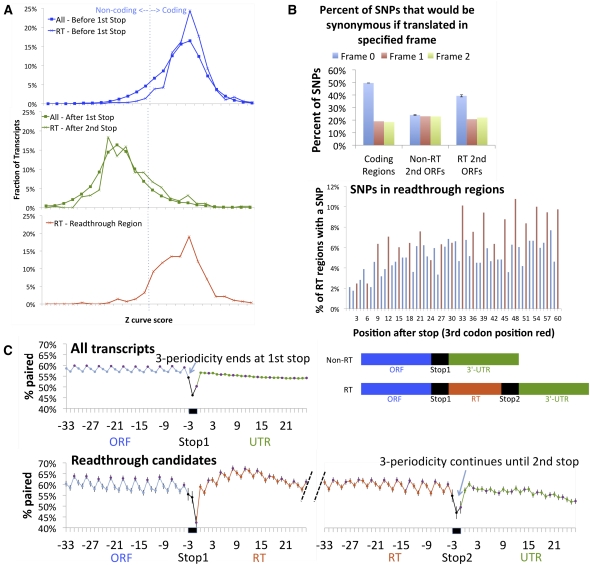
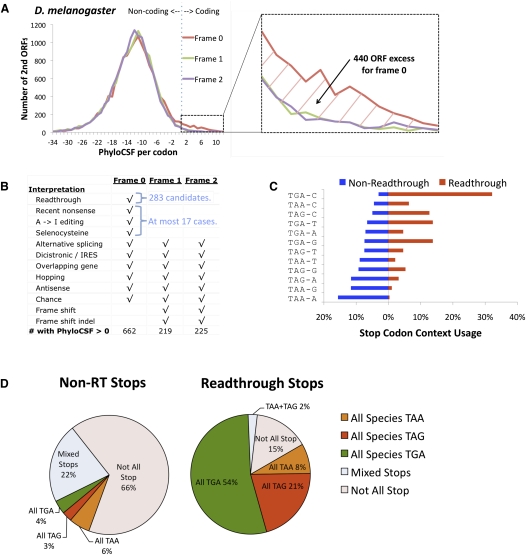
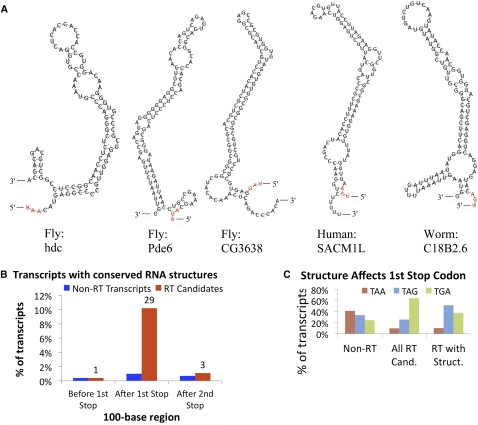
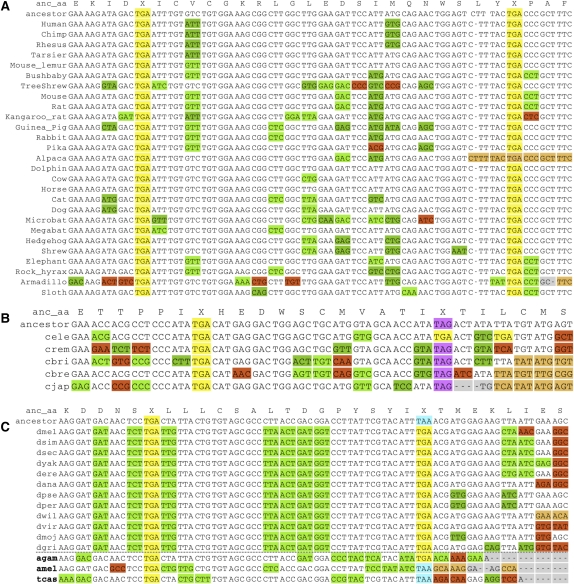
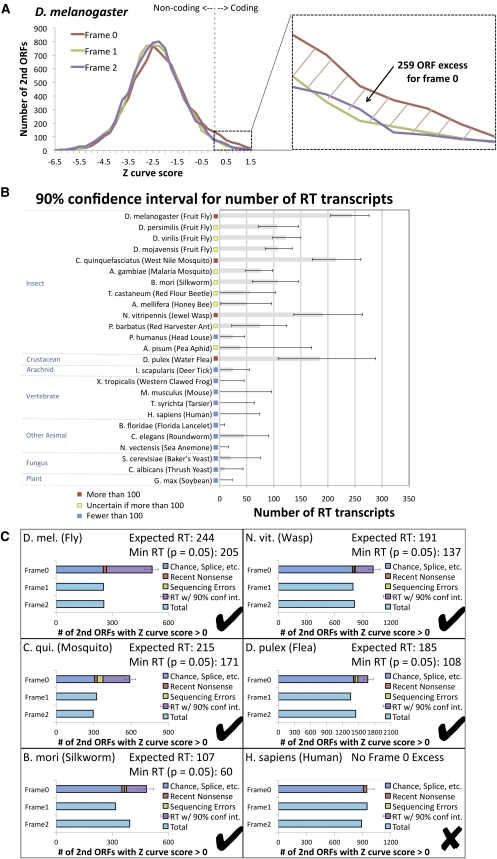
While translational stop codon readthrough is often used by viral genomes, it has been observed for only a handful of eukaryotic genes. We previously used comparative genomics evidence to recognize protein-coding regions in 12 species of Drosophila and showed that for 149 genes, the open reading frame following the stop codon has a protein-coding conservation signature, hinting that stop codon readthrough might be common in Drosophila. We return to this observation armed with deep RNA sequence data from the modENCODE project, an improved higher-resolution comparative genomics metric for detecting protein-coding regions, comparative sequence information from additional species, and directed experimental evidence. We report an expanded set of 283 readthrough candidates, including 16 double-readthrough candidates; these were manually curated to rule out alternatives such as A-to-I editing, alternative splicing, dicistronic translation, and selenocysteine incorporation. We report experimental evidence of translation using GFP tagging and mass spectrometry for several readthrough regions. We find that the set of readthrough candidates differs from other genes in length, composition, conservation, stop codon context, and in some cases, conserved stem-loops, providing clues about readthrough regulation and potential mechanisms. Lastly, we expand our studies beyond Drosophila and find evidence of abundant readthrough in several other insect species and one crustacean, and several readthrough candidates in nematode and human, suggesting that functionally important translational stop codon readthrough is significantly more prevalent in Metazoa than previously recognized.
Figures
References
-
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A 2004. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432: 112–118 - PubMed
-
- Aphasizhev R 2007. RNA editing. Mol Biol 41: 227–239
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases