

Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes
- PMID: 21994248
- PMCID: PMC3205576
- DOI: 10.1101/gr.108753.110
Locating protein-coding sequences under selection for additional, overlapping functions in 29 mammalian genomes
Abstract
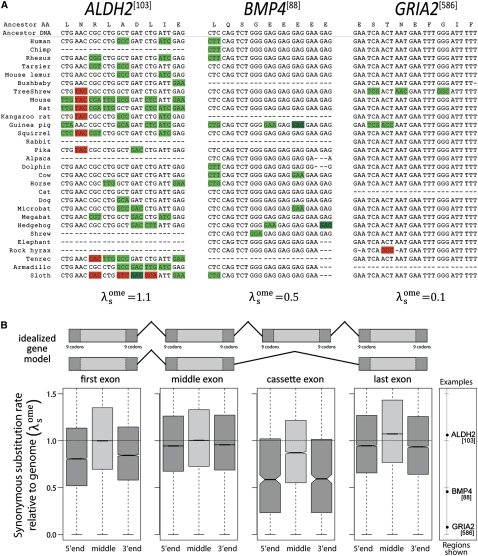
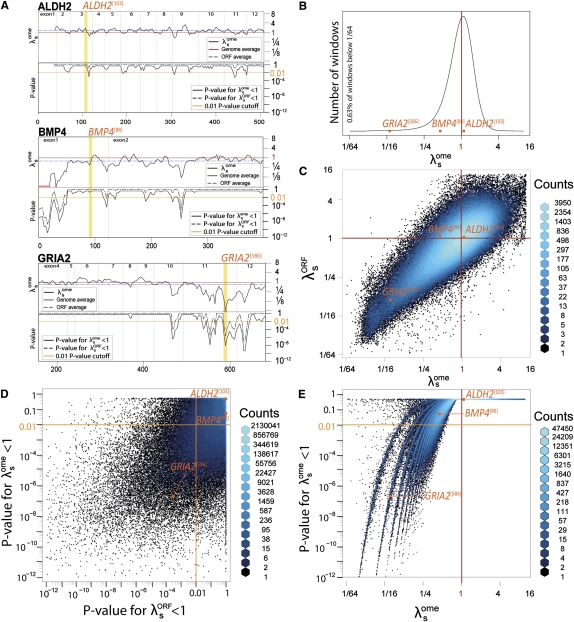
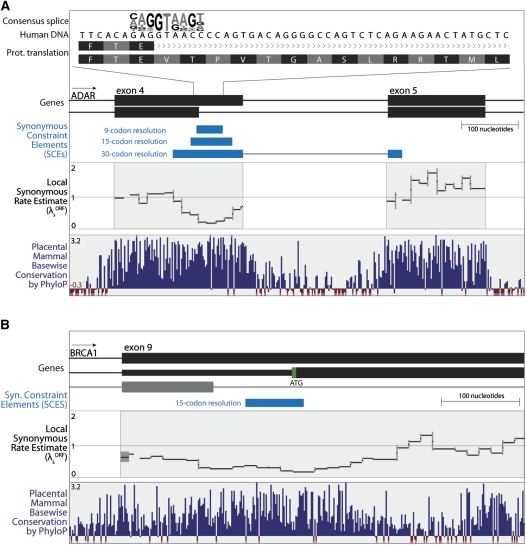
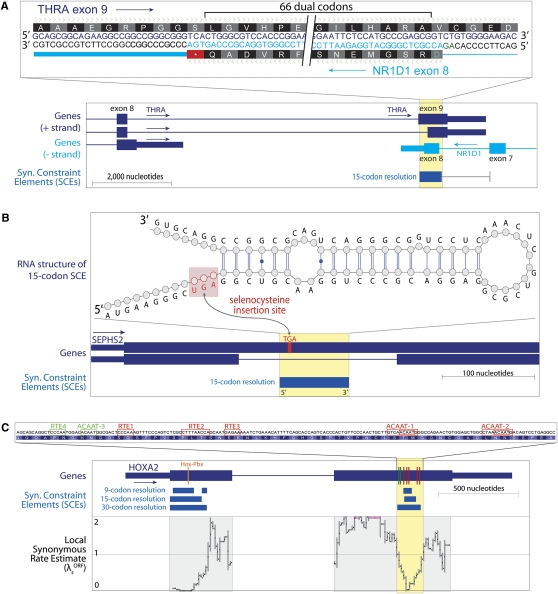
The degeneracy of the genetic code allows protein-coding DNA and RNA sequences to simultaneously encode additional, overlapping functional elements. A sequence in which both protein-coding and additional overlapping functions have evolved under purifying selection should show increased evolutionary conservation compared to typical protein-coding genes--especially at synonymous sites. In this study, we use genome alignments of 29 placental mammals to systematically locate short regions within human ORFs that show conspicuously low estimated rates of synonymous substitution across these species. The 29-species alignment provides statistical power to locate more than 10,000 such regions with resolution down to nine-codon windows, which are found within more than a quarter of all human protein-coding genes and contain ∼2% of their synonymous sites. We collect numerous lines of evidence that the observed synonymous constraint in these regions reflects selection on overlapping functional elements including splicing regulatory elements, dual-coding genes, RNA secondary structures, microRNA target sites, and developmental enhancers. Our results show that overlapping functional elements are common in mammalian genes, despite the vast genomic landscape.
Figures
Comment in
-
Comparative genomics: mammalian alignments reveal human functional elements.Nat Rev Genet. 2011 Nov 3;12(12):806-7. doi: 10.1038/nrg3112. Nat Rev Genet. 2011. PMID: 22048660 No abstract available.
References
-
- Anisimova M, Kosiol C 2009. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol 26: 255–271 - PubMed
-
- Anisimova M, Bielawski JP, Yang Z 2001. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol 18: 1585–1592 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources