

Estimation of rearrangement phylogeny for cancer genomes
- PMID: 21994251
- PMCID: PMC3266042
- DOI: 10.1101/gr.118414.110
Estimation of rearrangement phylogeny for cancer genomes
Abstract
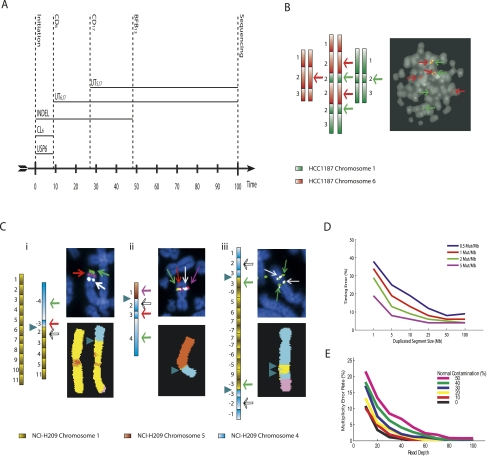
Cancer genomes are complex, carrying thousands of somatic mutations including base substitutions, insertions and deletions, rearrangements, and copy number changes that have been acquired over decades. Recently, technologies have been introduced that allow generation of high-resolution, comprehensive catalogs of somatic alterations in cancer genomes. However, analyses of these data sets generally do not indicate the order in which mutations have occurred, or the resulting karyotype. Here, we introduce a mathematical framework that begins to address this problem. By using samples with accurate data sets, we can reconstruct relatively complex temporal sequences of rearrangements and provide an assembly of genomic segments into digital karyotypes. For cancer genes mutated in rearranged regions, this information can provide a chronological examination of the selective events that have taken place.
Figures
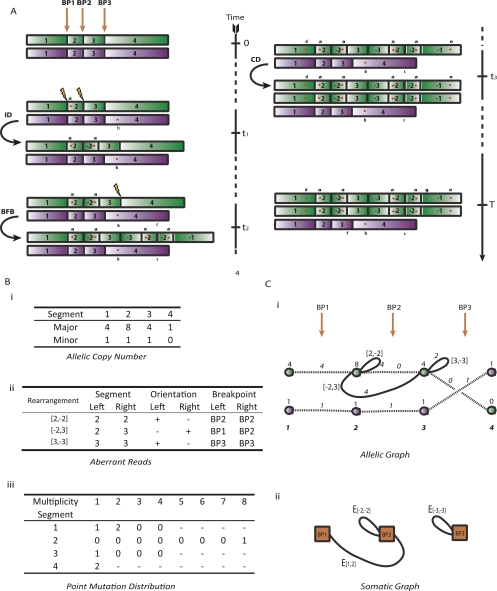
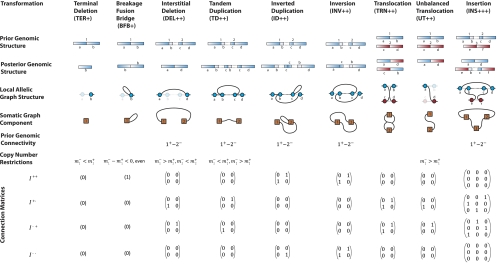
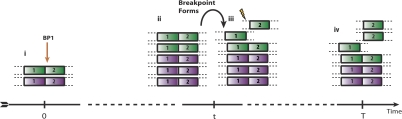
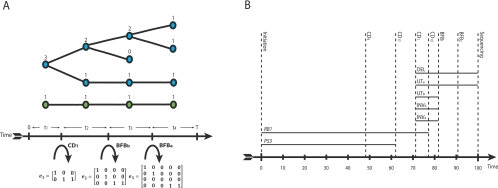
 represent copy numbers for segments to the left and right side of breakpoint i.
represent copy numbers for segments to the left and right side of breakpoint i.  indicates that the right side of breakpoint i must be genomically connected to the left side of breakpoint j prior to the transformation.
indicates that the right side of breakpoint i must be genomically connected to the left side of breakpoint j prior to the transformation.  indexes rearrangement between breakpoints i and j, where Si and Sj are the genomic orientations at the breakpoints.
indexes rearrangement between breakpoints i and j, where Si and Sj are the genomic orientations at the breakpoints.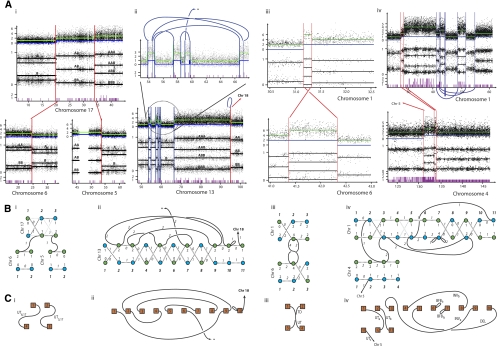
References
-
- Alekseyev MA, Pevzner PA 2007. Whole genome duplications and contracted breakpoint graphs. SIAM J Comput 36: 1748–1763
-
- Bader M, Ohlebusch E 2007. Sorting by weighted reversals, transpositions, and inverted transpositions. J Comput Biol 14: 615–636 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources