

The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity
- PMID: 21994366
- PMCID: PMC3213696
- DOI: 10.1523/JNEUROSCI.3059-11.2011
The melatonin MT1 receptor axis modulates mutant Huntingtin-mediated toxicity
Abstract
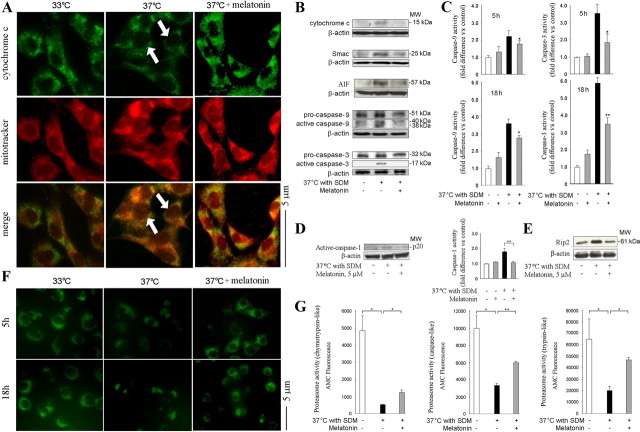
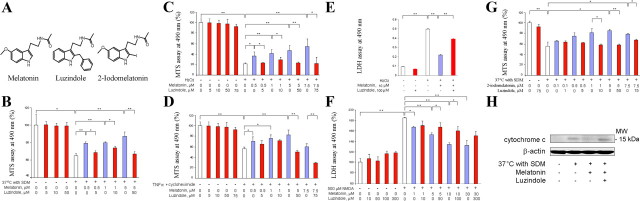
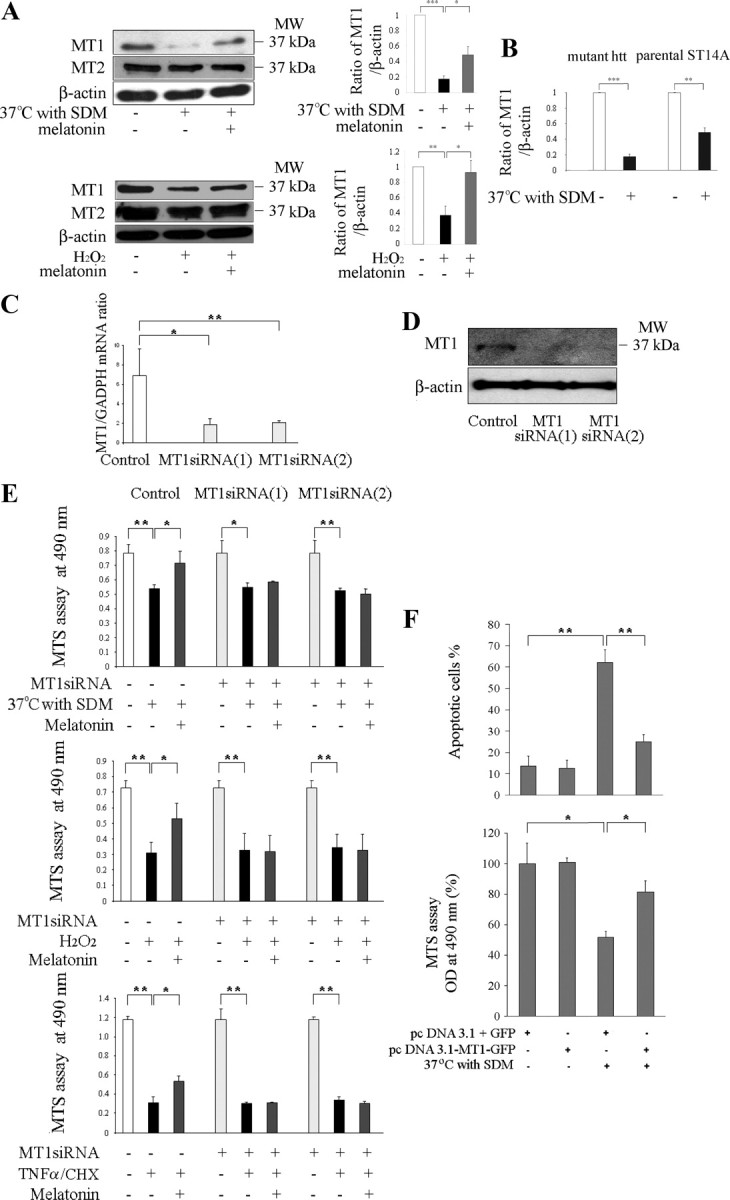
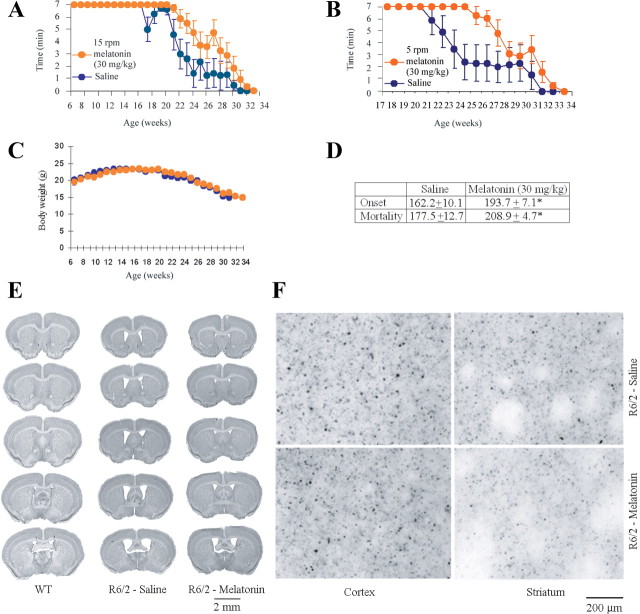
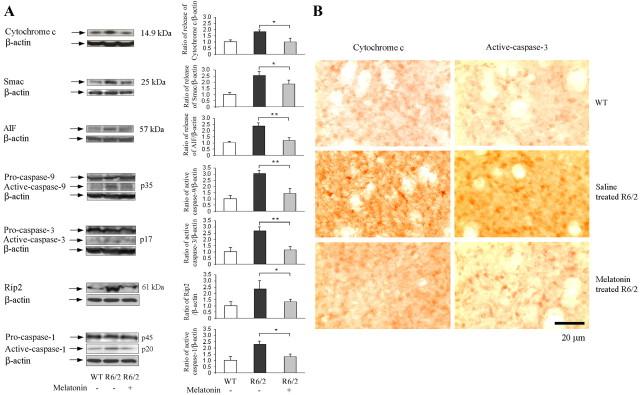
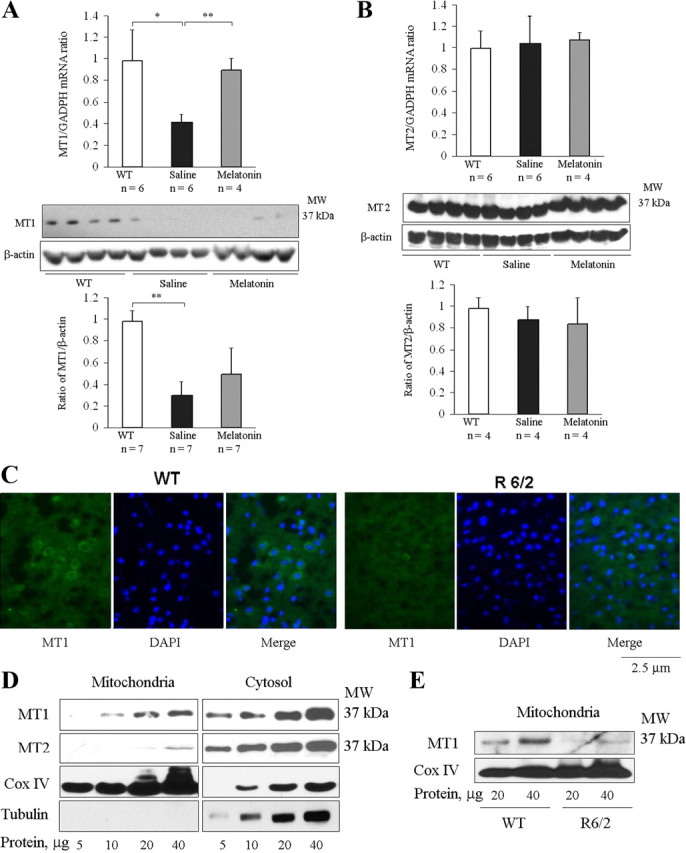
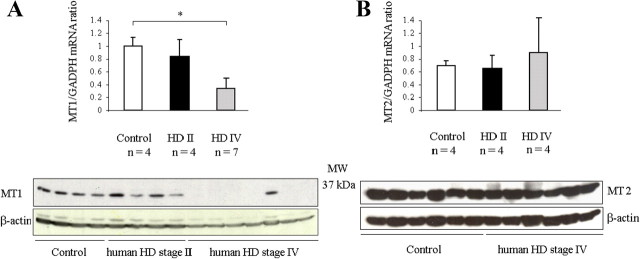
Melatonin mediates neuroprotection in several experimental models of neurodegeneration. It is not yet known, however, whether melatonin provides neuroprotection in genetic models of Huntington's disease (HD). We report that melatonin delays disease onset and mortality in a transgenic mouse model of HD. Moreover, mutant huntingtin (htt)-mediated toxicity in cells, mice, and humans is associated with loss of the type 1 melatonin receptor (MT1). We observe high levels of MT1 receptor in mitochondria from the brains of wild-type mice but much less in brains from HD mice. Moreover, we demonstrate that melatonin inhibits mutant htt-induced caspase activation and preserves MT1 receptor expression. This observation is critical, because melatonin-mediated protection is dependent on the presence and activation of the MT1 receptor. In summary, we delineate a pathologic process whereby mutant htt-induced loss of the mitochondrial MT1 receptor enhances neuronal vulnerability and potentially accelerates the neurodegenerative process.
Figures
References
-
- Adamczyk A, Czapski GA, Jesko H, Strosznajder RP. Non A beta component of Alzheimer's disease amyloid and amyloid beta peptides evoked poly(ADP-ribose) polymerase-dependent release of apoptosis-inducing factor from rat brain mitochondria. J Physiol Pharmacol. 2005;56(Suppl 2):5–13. - PubMed
-
- Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J. 2004;18:869–871. - PubMed
-
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. - PubMed
-
- Blazejová K, Nevsímalová S, Illnerová H, Hájek I, Sonka K. Sleep disorders and the 24-hour profile of melatonin and cortisol. Sb Lek. 2000;101:347–351. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical