

Orthopoxvirus genome evolution: the role of gene loss
- PMID: 21994715
- PMCID: PMC3185746
- DOI: 10.3390/v2091933
Orthopoxvirus genome evolution: the role of gene loss
Abstract
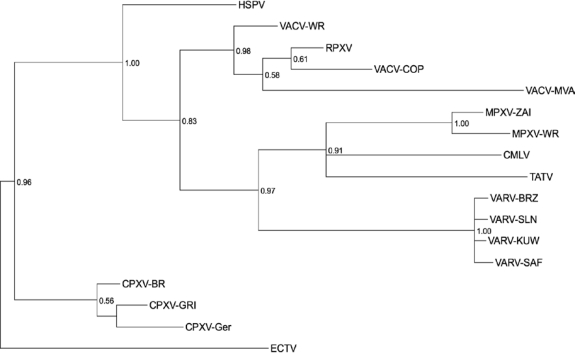
Poxviruses are highly successful pathogens, known to infect a variety of hosts. The family Poxviridae includes Variola virus, the causative agent of smallpox, which has been eradicated as a public health threat but could potentially reemerge as a bioterrorist threat. The risk scenario includes other animal poxviruses and genetically engineered manipulations of poxviruses. Studies of orthologous gene sets have established the evolutionary relationships of members within the Poxviridae family. It is not clear, however, how variations between family members arose in the past, an important issue in understanding how these viruses may vary and possibly produce future threats. Using a newly developed poxvirus-specific tool, we predicted accurate gene sets for viruses with completely sequenced genomes in the genus Orthopoxvirus. Employing sensitive sequence comparison techniques together with comparison of syntenic gene maps, we established the relationships between all viral gene sets. These techniques allowed us to unambiguously identify the gene loss/gain events that have occurred over the course of orthopoxvirus evolution. It is clear that for all existing Orthopoxvirus species, no individual species has acquired protein-coding genes unique to that species. All existing species contain genes that are all present in members of the species Cowpox virus and that cowpox virus strains contain every gene present in any other orthopoxvirus strain. These results support a theory of reductive evolution in which the reduction in size of the core gene set of a putative ancestral virus played a critical role in speciation and confining any newly emerging virus species to a particular environmental (host or tissue) niche.
Keywords: bioinformatics; evolution; orthopoxviruses; poxviruses; variola virus.
Figures

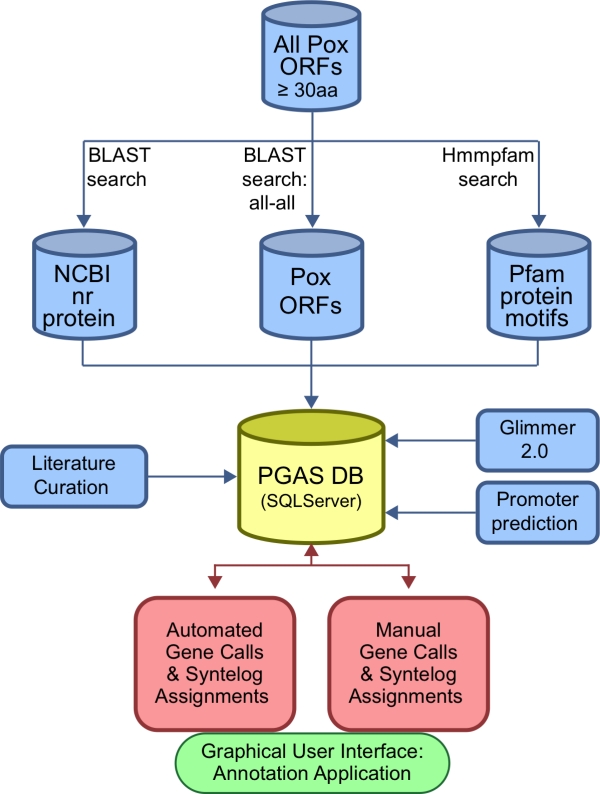



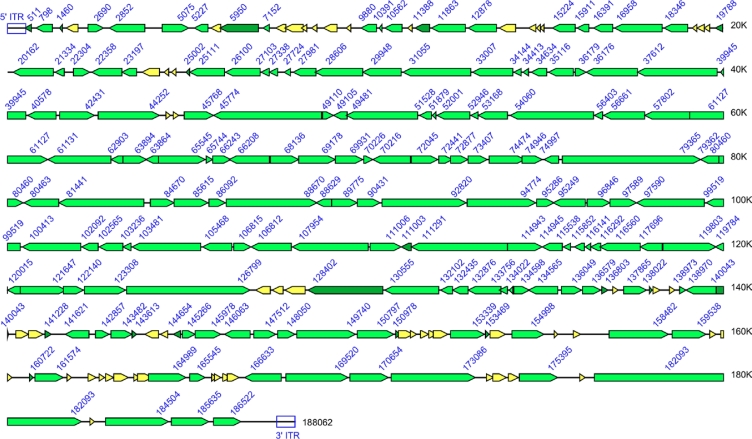
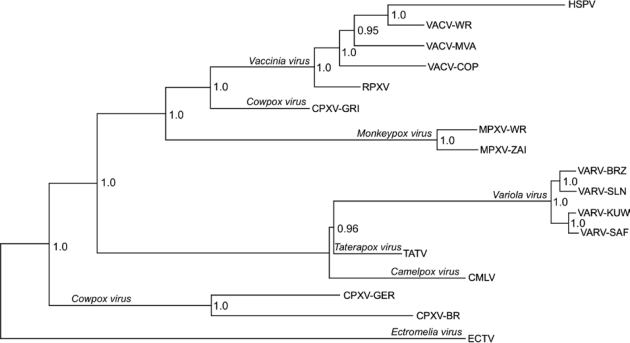

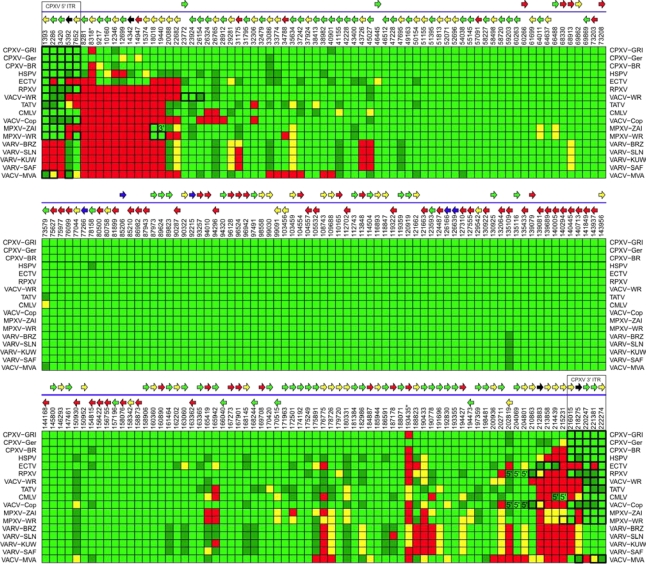

References
-
- Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. Virus Taxonomy: VIIIth Report of the International Committee on Taxonomy of Viruses. Academic Press (Elsevier); London, UK: 2005.
-
- Mercer AA, Schmidt A, Weber OF. Poxviruses. Birkhäuser Verlag; Basel, Switzerland: 2007.
-
- Barrett JW, McFadden G. Origin and evolution of poxviruses. In: Domingo E, Parrish CR, Holland JJ, editors. Origin and Evolution of Viruses. 2nd ed. Academic Press (Elsevier); London, UK: 2008. pp. 431–446.
-
- DeFilippis VR, Villarreal LP. Virus evolution. In: Knipe DM, Howley PM, editors. Fields Virology. 4th ed. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2001. pp. 353–370.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
