

The natural killer cell cytotoxic function is modulated by HIV-1 accessory proteins
- PMID: 21994772
- PMCID: PMC3185792
- DOI: 10.3390/v3071091
The natural killer cell cytotoxic function is modulated by HIV-1 accessory proteins
Abstract
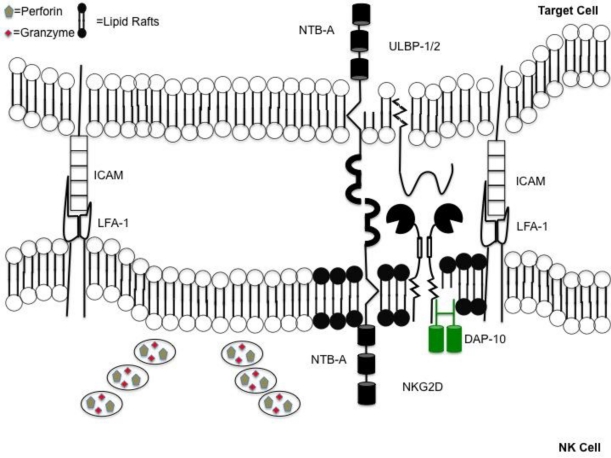
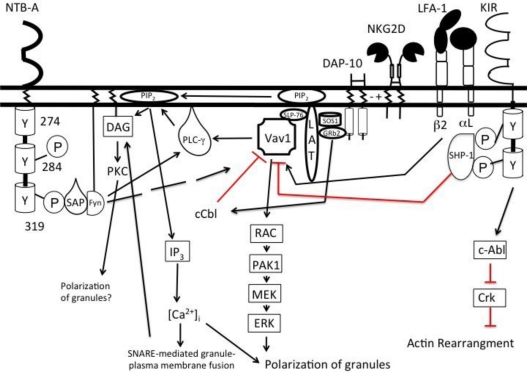
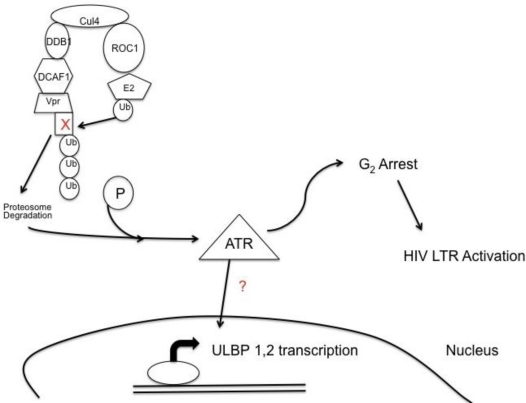
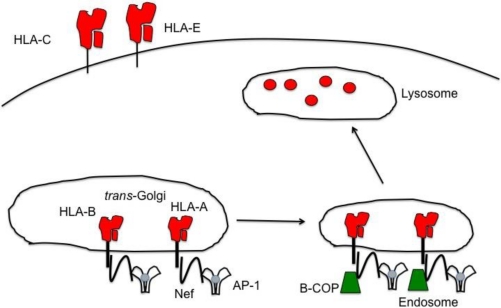
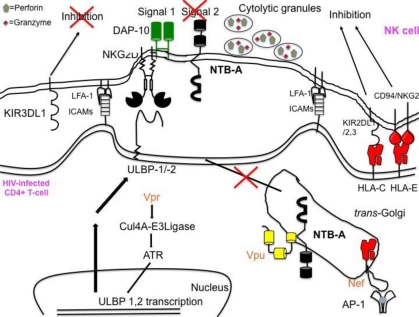
Natural killer (NK) cells' major role in the control of viruses is to eliminate established infected cells. The capacity of NK cells to kill virus-infected cells is dependent on the interactions between ligands on the infected cell and receptors on the NK cell surface. Because of the importance of ligand-receptor interactions in modulating the NK cell cytotoxic response, HIV has developed strategies to regulate various NK cell ligands making the infected cell surprisingly refractory to NK cell lysis. This is perplexing because the HIV-1 accessory protein Vpr induces expression of ligands for the NK cell activating receptor, NKG2D. In addition, the accessory protein Nef removes the inhibitory ligands HLA-A and -B. The reason for the ineffective killing by NK cells despite the strong potential to eliminate infected cells is due to HIV-1 Vpu's ability to down modulate the co-activation ligand, NTB-A, from the cell surface. Down modulation of NTB-A prevents efficient NK cell degranulation. This review will focus on the mechanisms through which the HIV-1 accessory proteins modulate their respective ligands, and its implication for NK cell killing of HIV-infected cells.
Keywords: HIV-1; NK cells; NKG2D; NTB-A; Nef; PLC-γ; Vpr; Vpu.
Figures
References
-
- Bukowski JF, Woda BA, Habu S, Okumura K, Welsh RM. Natural killer cell depletion enhances virus synthesis and virus-induced hepatitis in vivo. J. Immunol. 1983;131:1531–1538. - PubMed
-
- Habu S, Akamatsu K, Tamaoki N, Okumura K. In vivo significance of NK cell on resistance against virus (HSV-1) infections in mice. J. Immunol. 1984;133:2743–2747. - PubMed
-
- Stein-Streilein J, Guffee J, Fan W. Locally and systemically derived natural killer cells participate in defense against intranasally inoculated influenza virus. Reg. Immunol. 1988;1:100–105. - PubMed
-
- Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989;320:1731–1735. - PubMed
-
- Joncas J, Monczak Y, Ghibu F, Alfieri C, Bonin A, Ahronheim G, Rivard G. Brief report: killer cell defect persistent immunological abnormalities in two patients with chronic active Epstein-Barr virus infection. J. Med. Virol. 1989;28:110–117. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
