

Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ)
- PMID: 21994940
- PMCID: PMC3234872
- DOI: 10.1074/jbc.M111.311662
Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ)
Abstract
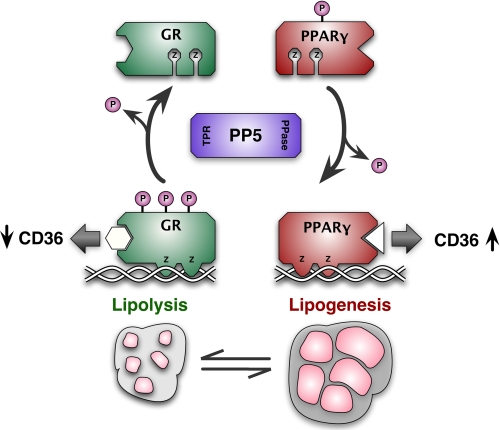
Glucocorticoid receptor-α (GRα) and peroxisome proliferator-activated receptor-γ (PPARγ) regulate adipogenesis by controlling the balance between lipolysis and lipogenesis. Here, we show that protein phosphatase 5 (PP5), a nuclear receptor co-chaperone, reciprocally modulates the lipometabolic activities of GRα and PPARγ. Wild-type and PP5-deficient (KO) mouse embryonic fibroblast cells were used to show binding of PP5 to both GRα and PPARγ. In response to adipogenic stimuli, PP5-KO mouse embryonic fibroblast cells showed almost no lipid accumulation with reduced expression of adipogenic markers (aP2, CD36, and perilipin) and low fatty-acid synthase enzymatic activity. This was completely reversed following reintroduction of PP5. Loss of PP5 increased phosphorylation of GRα at serines 212 and 234 and elevated dexamethasone-induced activity at prolipolytic genes. In contrast, PPARγ in PP5-KO cells was hyperphosphorylated at serine 112 but had reduced rosiglitazone-induced activity at lipogenic genes. Expression of the S112A mutant rescued PPARγ transcriptional activity and lipid accumulation in PP5-KO cells pointing to Ser-112 as an important residue of PP5 action. This work identifies PP5 as a fulcrum point in nuclear receptor control of the lipolysis/lipogenesis equilibrium and as a potential target in the treatment of obesity.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
