

Unique phenotype in a patient with CHARGE syndrome
- PMID: 21995344
- PMCID: PMC3216247
- DOI: 10.1186/1687-9856-2011-11
Unique phenotype in a patient with CHARGE syndrome
Abstract
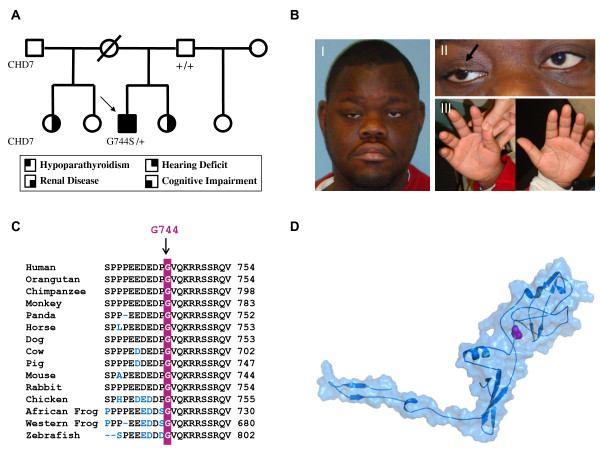
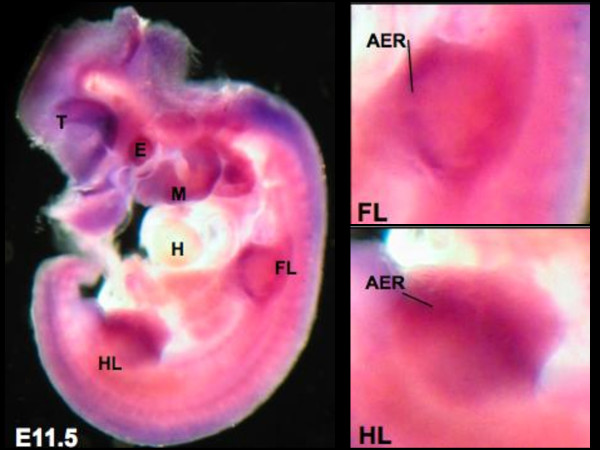
CHARGE is a phenotypically heterogeneous autosomal dominant disorder recognized as a cohesive syndrome since the identification of CHD7 as a genetic etiology. Classic features include: Coloboma, Heart defects, Atresia choanae, Retarded growth and development, Genitourinary abnormalities, and Ear anomalies and/or deafness. With greater accessibility to genetic analysis, a wider spectrum of features are emerging, and overlap with disorders such as DiGeorge syndrome, Kallmann syndrome, and Hypoparathyroidism Sensorineural Deafness and Renal Disease syndrome, is increasingly evident. We present a patient with a unique manifestation of CHARGE syndrome, including primary hypoparathyroidism and a limb anomaly; to our knowledge, he is also the first CHARGE subject reported with bilateral multicystic dysplastic kidneys. Furthermore, with structural modeling and murine expression studies, we characterize a putative CHD7 G744S missense mutation. Our report continues to expand the CHARGE phenotype and highlights that stringent fulfillment of conventional criteria should not strictly guide genetic analysis.
Figures
References
-
- Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BB, Brunner HG, Hoefsloot LH, van Ravenswaaij CM. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. Journal of medical genetics. 2006;43(4):306–14. - PMC - PubMed
-
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. American journal of human genetics. 2006;78(2):303–14. doi: 10.1086/500273. - DOI - PMC - PubMed
-
- Jongmans MC, Hoefsloot LH, van der Donk KP, Admiraal RJ, Magee A, van de Laar I, Hendriks Y, Verheij JB, Walpole I, Brunner HG, van Ravenswaaij CM. Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. American journal of medical genetics. 2008;146A(1):43–50. doi: 10.1002/ajmg.a.31921. - DOI - PubMed
-
- Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation--a syndrome. Journal of pediatric ophthalmology and strabismus. 1979;16(2):122–8. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
