

Bayesian hierarchical clustering for microarray time series data with replicates and outlier measurements
- PMID: 21995452
- PMCID: PMC3228548
- DOI: 10.1186/1471-2105-12-399
Bayesian hierarchical clustering for microarray time series data with replicates and outlier measurements
Abstract
Background: Post-genomic molecular biology has resulted in an explosion of data, providing measurements for large numbers of genes, proteins and metabolites. Time series experiments have become increasingly common, necessitating the development of novel analysis tools that capture the resulting data structure. Outlier measurements at one or more time points present a significant challenge, while potentially valuable replicate information is often ignored by existing techniques.
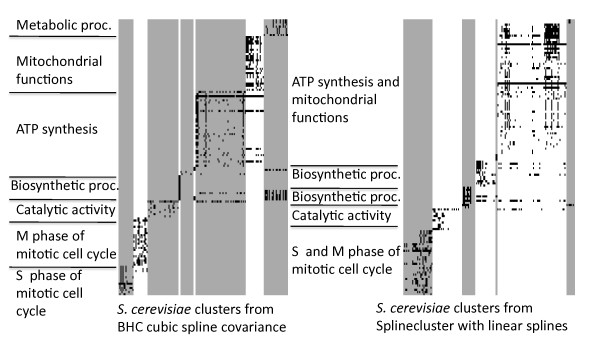
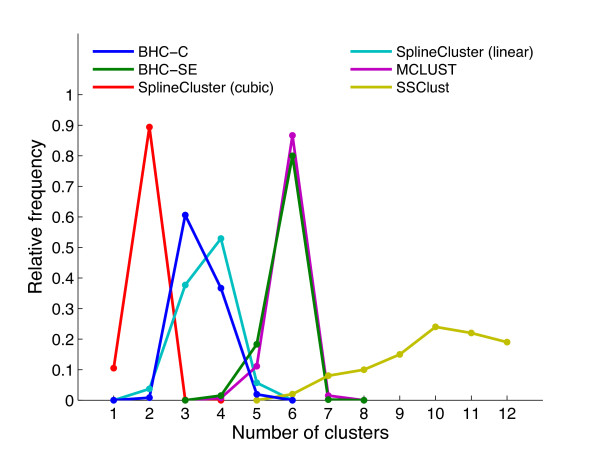
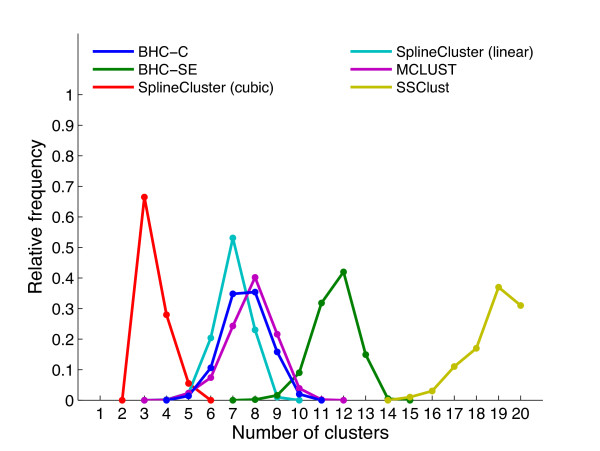
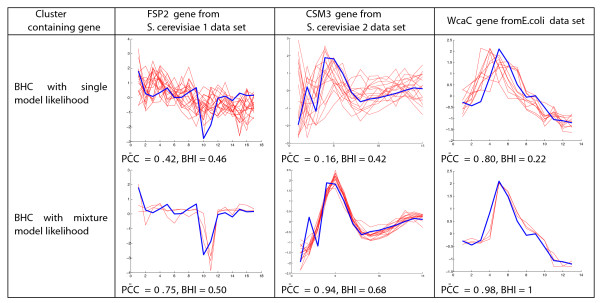
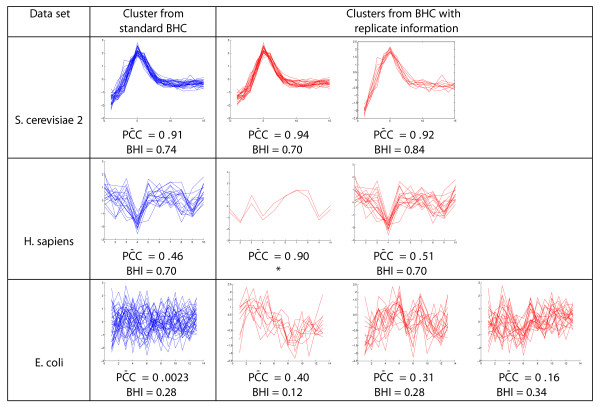
Results: We present a generative model-based Bayesian hierarchical clustering algorithm for microarray time series that employs Gaussian process regression to capture the structure of the data. By using a mixture model likelihood, our method permits a small proportion of the data to be modelled as outlier measurements, and adopts an empirical Bayes approach which uses replicate observations to inform a prior distribution of the noise variance. The method automatically learns the optimum number of clusters and can incorporate non-uniformly sampled time points. Using a wide variety of experimental data sets, we show that our algorithm consistently yields higher quality and more biologically meaningful clusters than current state-of-the-art methodologies. We highlight the importance of modelling outlier values by demonstrating that noisy genes can be grouped with other genes of similar biological function. We demonstrate the importance of including replicate information, which we find enables the discrimination of additional distinct expression profiles.
Conclusions: By incorporating outlier measurements and replicate values, this clustering algorithm for time series microarray data provides a step towards a better treatment of the noise inherent in measurements from high-throughput genomic technologies. Timeseries BHC is available as part of the R package 'BHC' (version 1.5), which is available for download from Bioconductor (version 2.9 and above) via http://www.bioconductor.org/packages/release/bioc/html/BHC.html?pagewanted=all.
Figures
References
-
- Beal M, Krishnamurthy P. Proceedings of the Proceedings of the Twenty-Second Conference Annual Conference on Uncertainty in Artificial Intelligence (UAI-06) Arlington, Virginia: AUAI Press; 2006. Gene Expression Time Course Clustering with Countably Infinite Hidden Markov Models; pp. 23–30.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
