

Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis
- PMID: 21995961
- PMCID: PMC3345964
- DOI: 10.1152/ajpgi.00301.2011
Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis
Abstract
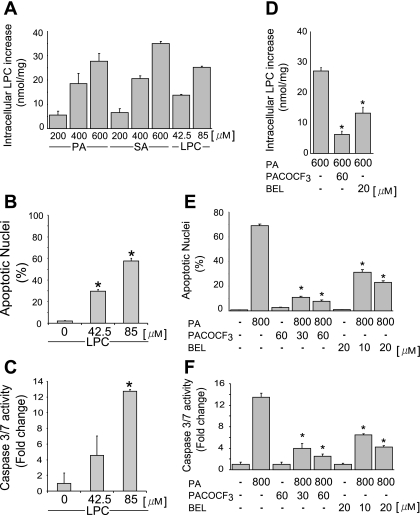
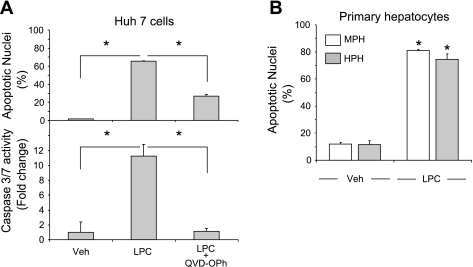
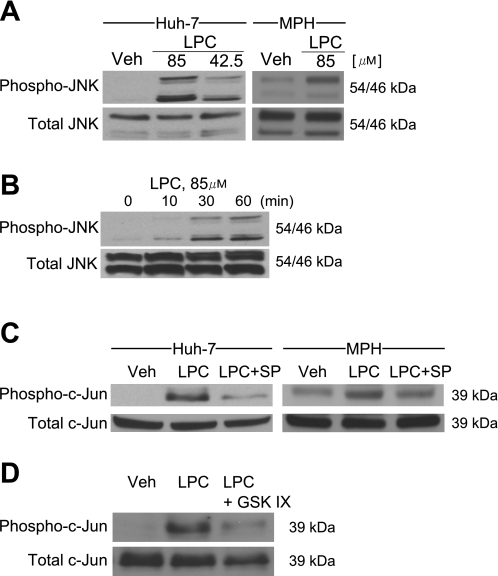
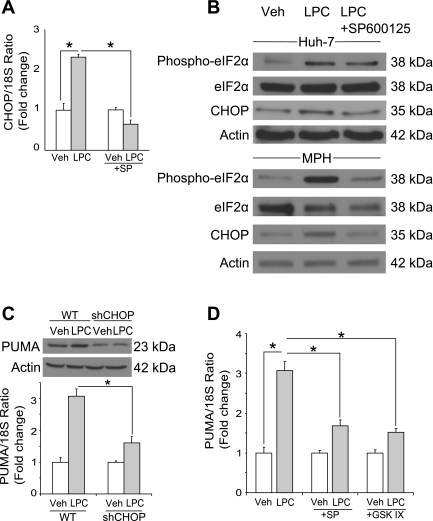
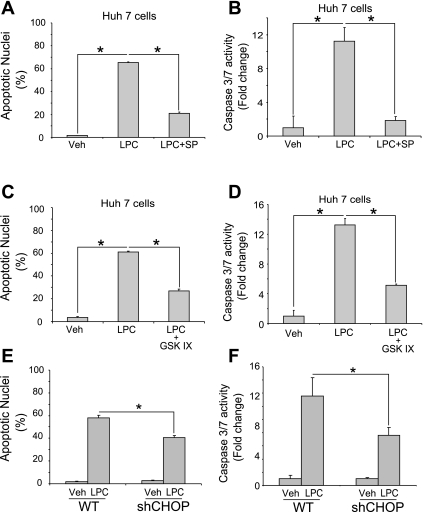
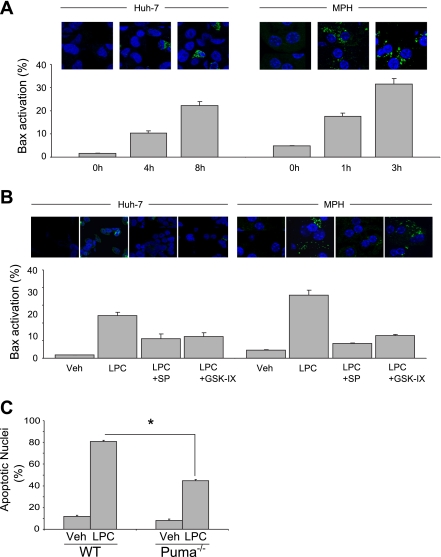
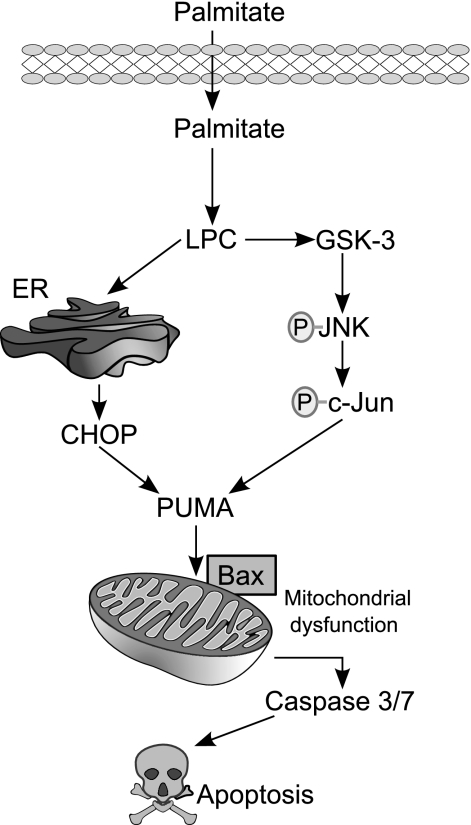
Isolated hepatocytes undergo lipoapoptosis, a feature of hepatic lipotoxicity, on treatment with saturated free fatty acids (FFA) such as palmitate (PA). However, it is unknown if palmitate is directly toxic to hepatocytes or if its toxicity is indirect via the generation of lipid metabolites such as lysophosphatidylcholine (LPC). PA-mediated hepatocyte lipoapoptosis is associated with endoplasmic reticulum (ER) stress, c-Jun NH(2)-terminal kinase (JNK) activation, and a JNK-dependent upregulation of the potent proapoptotic BH3-only protein PUMA (p53 upregulated modulator of apoptosis). Our aim was to determine which of these mechanisms of lipotoxicity are activated by PA-derived LPC. We employed Huh-7 cells and isolated murine and human primary hepatocytes. Intracellular LPC concentrations increase linearly as a function of the exogenous, extracellular PA, stearate, or LPC concentration. Incubation of Huh-7 cells or primary hepatocytes with LPC induced cell death by apoptosis in a concentration-dependent manner. Substituting LPC for PA resulted in caspase-dependent cell death that was accompanied by activating phosphorylation of JNK with c-Jun phosphorylation and an increase in PUMA expression. LPC also induced ER stress as manifest by eIF2α phosphorylation and CAAT/enhancer binding homologous protein (CHOP) induction. LPC cytotoxicity was attenuated by pharmacological inhibition of JNK or glycogen synthase kinase-3 (GSK-3). Similarly, short-hairpin RNA (shRNA)-targeted knockdown of CHOP protected Huh-7 cells against LPC-induced toxicity. The LPC-induced PUMA upregulation was prevented by JNK inhibition or shRNA-targeted knockdown of CHOP. Finally, genetic deficiency of PUMA rendered murine hepatocytes resistant to LPC-induced apoptosis. We concluded that LPC-induced lipoapoptosis is dependent on mechanisms largely indistinguishable from PA. These data suggest that FFA-mediated cytotoxicity is indirect via the generation of the toxic metabolite, LPC.
Figures
References
-
- Adams LA, Lymp JF, Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129: 113–121, 2005 - PubMed
-
- Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol 17, Suppl: S186–S190, 2002 - PubMed
-
- Ariyama Y, Tanaka Y, Shimizu H, Shimomura K, Okada S, Saito T, Yamada E, Oyadomari S, Mori M. The role of CHOP messenger RNA expression in the link between oxidative stress and apoptosis. Metabolism 57: 1625–1635, 2008 - PubMed
-
- Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem 282: 27141–27154, 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous