

Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis
- PMID: 21997559
- PMCID: PMC3539739
- DOI: 10.1136/gutjnl-2011-301008
Oncogenic transcription factors: cornerstones of inflammation-linked pancreatic carcinogenesis
Abstract
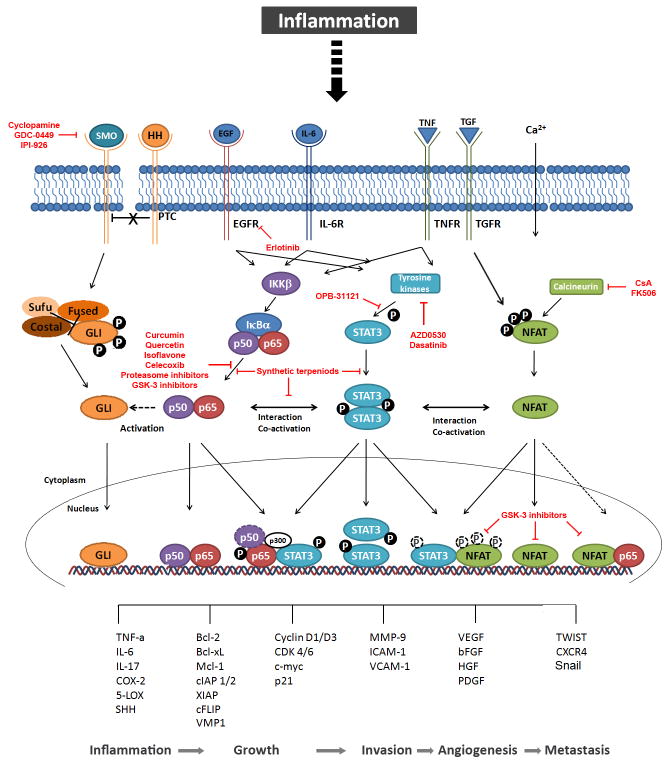
Transcription factors are proteins that regulate gene expression by modulating the synthesis of messenger RNA. Since this process is often one dominant control point in the production of many proteins, transcription factors represent the key regulators of numerous cellular functions, including proliferation, differentiation and apoptosis. Pancreatic cancer progression is characterised by activation of inflammatory signalling pathways converging on a limited set of transcription factors that fine-tune gene expression patterns contributing to the growth and maintenance of these tumours. Thus strategies targeting these transcriptional networks activated in pancreatic cancer cells could block the effects of upstream inflammatory responses participating in pancreatic tumorigenesis. The authors review this field of research and summarise current strategies for targeting oncogenic transcription factors and their activating signalling networks in the treatment of pancreatic cancer.
Figures
References
-
- Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. - PubMed
-
- Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. - PubMed
-
- Erkan M, Reiser-Erkan C, Michalski CW, et al. Tumor microenvironment and progression of pancreatic cancer. Exp Oncol. 2010;32:128–131. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical